Aldostérone et cœur M Aldosterone and the heart

Mise au point
Mise au point
19
La Lettre du Cardiologue - n° 405 - mai 2007
Aldostérone et cœur1
Aldosterone and the heart
IPR.C. Gaillard*
1 © Métabolismes Hormones Diabètes et Nutrition (X), n° 6, novembre-décembre 2006.
* Service d’endocrinologie, diabétologie et métabolisme, CHU Vaudois, 1011 Lausanne, Suisse.
RÉSUMÉ
Le cœur est non seulement un organe cible pour les actions
hormonales mais aussi un organe endocrine qui synthé-
tise et sécrète des hormones. Ainsi le cœur est-il à la fois
un organe cible de l’aldostérone et de l’angiotensine II,
dont il est aussi une source. Il produit de l’angiotensine II
soit par la voie classique du système rénine-angiotensine-
aldostérone, dépendante de l’enzyme de conversion de
l’angiotensine (ECA), soit par une voie alterne indépen-
dante de cette dernière, la voie de la chymase (une sérine
protéinase présente dans les mastocytes). Outre ses eff ets
classiques qui assurent l’homéostasie hydro-électrolytique,
l’aldostérone induit de nombreux eff ets délétères (fi brose
myocardique, dysfonction endothéliale, augmentation du
PAI-1, etc.) responsables d’une augmentation de la morbi-
dité et de la mortalité cardiovasculaires. Associé au traite-
ment conventionnel, le blocage spécifi que du récepteur
minéralocorticoïde par de faibles doses de spironolactone
ou d’éplérénone réduit de manière très signifi cative cette
morbi-mortalité chez des patients atteints d’une insuffi -
sance cardiaque modérée à sévère et/ou d’une dysfonction
ventriculaire gauche après infarctus du myocarde.
Mots-clés : Système rénine-angiotensine-aldostérone –
Chymase mastocytaire – Fibrose myocardique – Arythmie –
Récepteur minéralocorticoïde – Spironolactone – Épléré-
none.
Keywords: Renin-angiotensin-aldosterone system – Mast
cells chymase – Myocardial fibrosis – Arrhythmia – Mineralo-
corticoid receptor – Spironolactone – Eplerenone.
Le cœur est non seulement un organe cible pour les
actions directes et indirectes de multiples hormones
et facteurs hormonaux, mais il constitue également
un organe endocrine, au sens classique du terme, puisqu’il
synthétise et sécrète dans la circulation des substances qui
vont agir à distance.
Après la découverte du facteur natriurétique auriculaire (ANF)
dans les années 1980, il a été montré que le cœur fonctionnait
comme un véritable organe endocrine. En eff et, De Bold et al. (1)
ont démontré en 1981 que l’injection à des animaux de laboratoire
d’extraits cardiaques d’origine auriculaire, mais non d’origine
ventriculaire, induisait une natriurèse et une réduction du volume
intravasculaire. Cette activité fut attribuée à l’ANF, qui, après son
isolement et sa caractérisation, est devenu le peptide natriuré-
tique auriculaire (ANP). Celui-ci est synthétisé par le cœur, d’où
il est sécrété dans la circulation pour agir au niveau rénal, où il
se comporte comme un puissant agent natriurétique, ainsi qu’au
niveau vasculaire, où il induit une vasodilatation. L’ANP agit égale-
ment sur la contractilité du myocarde. Depuis la découverte de
l’ANP, d’autres peptides ou facteurs humoraux cardiaques ont été
identifi és. Parmi eux, citons l’adrénomédulline, dont la synthèse
est stimulée par une surcharge de pression et de volume (2), et
l’urocortine, un nouveau membre des peptides de la famille du
CRH (3). Ces deux peptides sont de puissants vasodilatateurs.
En outre, l’adrénomédulline diminue le remodelage vasculaire
par un eff et antiprolifératif au niveau de la paroi vasculaire, et
pourrait ainsi exercer une protection contre le développement
de l’athérosclérose et/ou de la resténose (4, 5). Le cœur est égale-
ment capable, comme nous le mentionnerons dans cette brève
revue, de produire lui-même tous les composants du système
rénine-angiotensine-aldostérone (SRAA). Il produit encore de
nombreux autres facteurs humoraux tels que le monoxyde d’azote
(NO), l’endothéline, les éicosanoïdes et les substances du système
kallicréine-kinine. Toutes ces substances sont des modulateurs
de la performance cardiaque, que ce soit dans des circonstances
physiologiques ou physiopathologiques (6).
Le cœur constitue aussi un organe cible pour des hormones
et certains facteurs humoraux, comme les catécholamines, les
substances du SRAA, les estrogènes, la testostérone, l’insuline,
ainsi que pour l’hormone de croissance et les sécrétines peptidi-
ques de l’hormone de croissance (6-9). Ainsi, dans l’insuffi sance
cardiaque, le cœur est incapable d’assurer correctement sa fonc-
tion de pompe, et la baisse du débit cardiaque qui en résulte
entraîne une augmentation des concentrations plasmatiques de
plusieurs substances actives au niveau cardiovasculaire, telles les
catécholamines, l’angiotensine II, l’aldostérone, la vasopressine
et l’endothéline. L’action de toutes ces substances a pour but de
maintenir la pression de perfusion périphérique. À long terme,
toutefois, les actions de ces hormones se révèlent dommageables,
car elles sont responsables d’eff ets délétères sur la structure et
la fonction cardiaques, et contribuent ainsi paradoxalement à
l’aggravation de la dysfonction cardiaque. L’activation de ces
systèmes neurohormonaux sert de marqueur de l’insuffi sance
cardiaque et constitue ainsi une cible thérapeutique. Parmi ces

Mise au point
Mise au point
20
La Lettre du Cardiologue - n° 405 - mai 2007
hormones, il a été récemment démontré que les composants du
SRAA ont eff ectivement des eff ets spécifi ques et néfastes sur le
cœur et les vaisseaux, et que le blocage de leur action pourrait
ainsi être bénéfi que.
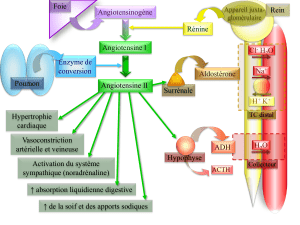
LE SYSTÈME RÉNINEANGIOTENSINEALDOSTÉRONE
Le SRAA préserve l’homéostasie circulatoire lors d’une perte
de sel et d’eau résultant par exemple d’une transpiration intense
et prolongée, de vomissements ou d’une diarrhée. La rénine,
l’angiotensine II et l’aldostérone sont des éléments clés de ce
système. La rénine, synthétisée par l’appareil juxtaglomérulaire
du rein, produira le clivage de quatre acides de l’angiotensino-
gène circulant (le précurseur hépatique de tous les peptides de
l’angiotensine) pour former l’angiotensine I, un décapeptide
biologiquement inactif. L’enzyme de conversion de l’angioten-
sine (ECA) qui est liée à la membrane des cellules endothéliales
eff ectue ensuite le clivage de deux acides aminés de l’angioten-
sine I pour engendrer l’angiotensine II.
La chymase mastocytaire
L’ECA est donc une enzyme indispensable à la génération de
l’angiotensine II. Dans le cœur, il existe cependant une voie
alterne ne passant pas par l’ECA, mais impliquant l’α-chymase,
une enzyme similaire à la chymotrypsine qui est exprimée dans
les granules sécrétoires des mastocytes cardiaques. La présence
d’une telle voie a été démontrée in vitro, puisque, dans des
homogénats de tissu cardiaque humain, 75 % de l’angiotensine II
formée sont dépendants de la chymase, alors que seuls 25 % le
sont de la voie classique de l’ECA (10, 11). Diverses études ont
confi rmé que l’angiotensine II, dans les divers tissus d’origine
cardiovasculaire, est dépendante de la voie de la chymase plutôt
que de la voie classique de l’ECA, puisque la production d’an-
giotensine II dans ces tissus est bloquée à près de 90 % par des
inhibiteurs de la chymase (12-14).
La chymase est stockée dans les granules des mastocytes sous
forme inactive, en raison d’un pH à 5,5 dans ces granules. Le
pH optimal pour l’activité enzymatique est compris entre 7
et 9, situation rencontrée lors de la sécrétion de la chymase
après activation des mastocytes présents dans un tissu lésé ou
infl ammatoire (15). L’activité chymasique ne s’exerce qu’au niveau
tissulaire local, car de puissants inhibiteurs de la chymase sont
présents dans la circulation sanguine et bloquent immédiatement
son activité enzymatique (15).
Il existe deux types de chymase, la forme α, qui prédomine
notamment chez l’homme, le singe et le mouton, et la forme β,
que l’on trouve chez le rat et la souris (16). La chymase cardiaque
humaine présente une très grande spécifi cité pour l’angioten-
sine I, ce qui la distingue d’autres enzymes impliquées dans la
production de l’angiotensine II, telles l’ECA, la kallicréine, la
cathepsine et les autres chymases (10). En outre, la chymase
cardiaque humaine, au contraire de l’ECA, ne dégrade pas l’an-
giotensine II ni les autres peptides comme la bradykinine, la
substance P et la gonadolibérine (10).
La chymase mastocytaire contribue non seulement à la transfor-
mation de l’angiotensine I en angiotensine II, mais elle participe
aussi à la sécrétion et à l’activation de la cytokine IL-1β, une
cytokine pro-infl ammatoire, et du TGF-β1, une cytokine qui est
impliquée notamment dans l’hypertrophie et la fi brose cardiaque
(17, 18). La chymase active le TGF-β qui se trouve sous forme
inactive ou latente dans les mastocytes (18). Les mastocytes
sont donc, par ce mécanisme, impliqués dans des processus
pathologiques caractérisés par des événements infl ammatoires
et fi brogéniques tels que la fi brose pulmonaire (19), l’infarctus
du myocarde et la fi brose myocardique post-transplantation
(20, 21). Toutes ces observations suggèrent qu’une inhibition
de ces eff ets par des inhibiteurs de la chymase pourrait être
utilisée à but thérapeutique pour prévenir les maladies cardio-
vasculaires et la fi brose.
En plus des eff ets susmentionnés, l’α-chymase est aussi capable
de former les endothélines de 31 acides aminés, de dégrader
l’endothéline 1, de modifi er le métabolisme lipidique et d’en-
dommager la matrice extracellulaire (22).
L’angiotensine II exerce de nombreuses actions pour maintenir
l’homéostasie circulatoire, induisant la constriction des arté-
rioles au niveau des circulations rénale et systémique, ainsi que
la réabsorption du sodium au niveau des segments proximaux
du néphron. L’angiotensine II stimule également la sécrétion
d’aldostérone responsable d’une réabsorption du sodium (en
échange du potassium) au niveau des segments distaux du néphron,
ainsi que dans le côlon, les glandes salivaires et sudoripares.
Lorsque le volume intravasculaire est réduit, l’angiotensine II
est le stimulus principal de la production d’aldostérone, qui peut
cependant aussi être stimulée par le potassium, l’ACTH, les
catécholamines, l’endothéline et la sérotonine d’origine masto-
cytaire. Parmi ces stimuli de l’aldostérone, le potassium a un
rôle physiologique important, puisqu’il permet de maintenir son
homéostasie grâce à la capacité de l’aldostérone, dont il stimule
la sécrétion, à augmenter l’excrétion potassique dans les urines,
les selles, la sueur et la salive (23). L’aldostérone permet ainsi
de prévenir l’hyperkaliémie en cas d’apport potassique élevé
dans l’alimentation ou après un exercice physique intense qui
entraîne une sortie de potassium des muscles.
L’aldostérone produit deux types d’action : des modifi cations
immédiates de conductances ioniques, ainsi que l’induction
diff érée de gènes cibles qui survient après liaison sur son récep-
teur intracellulaire, le récepteur des minéralocorticoïdes (MR).
Son action principale consiste à augmenter la synthèse et l’acti-
vité des canaux sodiques amiloride-sensibles et la Na/K-ATPase
membranaire des cellules du tubule contourné distal et du tube
collecteur rénal pour aboutir à la réabsorption du sodium.
Comme nous le verrons ci-après, l’aldostérone, parallèlement
à cette action essentielle de réabsorption sodique, exerce d’autres
actions, notamment sur le système cardiovasculaire.
Le récepteur MR de l’aldostérone est un facteur de transcrip-
tion, membre de la superfamille des récepteurs nucléaires, qui
comprend les récepteurs aux hormones stéroïdes/thyroïdes et à
l’acide rétinoïque. Ces récepteurs sont classiquement localisés
dans des cellules épithéliales (côlon, reins, glandes salivaires

Mise au point
Mise au point
21
La Lettre du Cardiologue - n° 405 - mai 2007
et sudoripares). Des études récentes ont toutefois démontré
que les MR pouvaient également être présents dans des tissus
non épithéliaux tels que les organes circumventriculaires du
cerveau, le cœur et les vaisseaux sanguins. Les domaines de
fi xation sur l’ADN et de liaison au ligand du MR et du récep-
teur des glucocorticoïdes (GR) ont une grande homologie
de séquences. Pour cette raison, le MR peut également lier
le cortisol avec la même affi nité que pour l’aldostérone. Le
cortisol peut être un véritable concurrent pour l’aldostérone,
car sa concentration plasmatique est 100 à 1 000 fois supé-
rieure à celle de l’aldostérone. Aussi, pour “protéger” le MR des
glucocorticoïdes, les cellules disposent-elles d’une enzyme, la
11-β-hydroxystéroïde deshydrogénase (11-β-HSD) de type 2,
qui transforme le cortisol (actif) en cortisone, inactive car
incapable de se lier au MR. Cela suppose donc que, dans les
types cellulaires spécifi quement répondeurs à l’aldostérone,
il y ait une colocalisation de la 11-β-HSD 2 et du MR. Une
telle colocalisation a été vérifi ée dans le rein, et elle est aussi
présente dans le cœur humain (24).
SYNTHÈSE DE L’ALDOSTÉRONE
La synthèse de l’aldostérone a lieu dans le cortex surrénalien, où
la P-450 aldostérone-synthase, sous contrôle de l’angiotensine II
et du potassium, et plus faiblement de l’ACTH et du sodium,
catalyse cette synthèse à partir de la désoxycorticostérone.
Des sites de production extrasurrénaliens ont cependant été
identifi és. Il s’agit du cœur, de l’aorte, de l’artère pulmonaire
et du cerveau. Un SRAA complet a été mis en évidence au
niveau du cœur (25), qui possède ainsi l’ensemble de l’équi-
pement enzymatique nécessaire à la production d’aldostérone
(26). Le cœur est également un organe cible pour les eff ets
de l’aldostérone, puisque le messager du MR est abondant
dans les cardiomyocytes et la paroi endothéliale des artères
principales (24). La concentration tissulaire cardiaque d’aldos-
térone, qui est déjà 15 fois supérieure à celle du plasma, peut
encore augmenter de deux à trois fois dans le modèle murin de
situation postinfarctus par ligature coronaire (27). Toutefois,
la contribution cardiaque aux taux plasmatiques d’aldostérone
paraît non signifi cative comparativement à celle du cortex
surrénalien (28). Si elle ne contribue pas aux taux circulants
de cette hormone, elle joue certainement un rôle paracrine
et autocrine important au niveau cardiaque. Il est intéressant
de noter que le sel a une fonction régulatrice diff érente sur
l’aldostérone plasmatique et sur l’aldostérone cardiaque. En
eff et, un régime hypersodé chez le rat normotendu induit
une diminution de l’aldostérone plasmatique, alors qu’une
augmentation de l’aldostérone cardiaque et une hypertrophie
du cœur sont relevées (29). Cette observation suggère donc
qu’un régime hypersodé augmente la synthèse d’aldostérone du
cœur et contribue ainsi à l’hypertrophie cardiaque de manière
indépendante du SRAA circulant. Des travaux récents ont
clairement démontré que l’aldostérone exerçait des eff ets
directs sur le cardiomyocyte, par l’induction, en présence d’une
concentration extracellulaire de sodium, d’une hypertrophie
cardiaque (30). De plus, au niveau du cœur, l’aldostérone exerce
un rétrocontrôle positif sur sa propre production, puisqu’elle
augmente paradoxalement l’expression de l’ARN messager de
l’enzyme de conversion. Ce mécanisme pourrait être respon-
sable de l’augmentation et de la progression de l’activité du
SRAA cardiaque en cas d’insuffi sance cardiaque.
Le cœur est donc non seulement un organe cible pour les miné-
ralocorticoïdes et les glucocorticoïdes, mais il semble également
constituer un organe endocrine capable de synthétiser de novo
des corticostéroïdes. Il y a cependant controverse quant à l’ori-
gine précise de l’aldo-stérone cardiaque (31-33), des arguments
scientifi ques solides amenant à considérer que l’aldostérone
intracardiaque provient essentiellement de la circulation géné-
rale (28).
EFFETS DE L’ALDOSTÉRONE
Les eff ets minéralocorticoïdes classiques incluent la rétention
du sodium et de l’eau ainsi que l’excrétion du potassium et du
magnésium. Ils contrôlent l’homéostasie du sel et de l’eau.
Depuis quelques années, de nouveaux eff ets ont été mis en
évidence, en particulier au niveau du cœur et des vaisseaux. Ces
derniers sont plutôt considérés comme des eff ets indésirables.
Ils sont responsables de l’induction d’une fi brose cardiaque
importante, d’une fi brose/rigidité vasculaire, d’une diminution
de la capture myocardique de norépinéphrine, d’une augmen-
tation de la production de l’inhibiteur de l’activateur du plas-
minogène de type 1 (PAI-1), de l’induction d’une dysfonction
endothéliale, et d’une diminution du HDL-cholestérol. Autant
d’eff ets dont les conséquences délétères sont résumées dans
le tableau.
Tableau.
Conséquences délétères de l’augmentation de l’aldostérone.
Rétention sodée Insuffi sance cardiaque
Perte potassique Arythmies ventriculaires
Perte de magnésium Toxicité digitalique
Fibrose myocardique
Production de collagène du myocarde Dysfonction ventriculaire
Hypertrophie ventriculaire
Diminution de la réserve coronarienne Ischémie
Dysfonction des barorécepteurs Hypertension artérielle
Diminution capture myocardique NE Arythmies
Dysfonction endothéliale ATS
Diminution du HDL-cholestérol ATS
Augmentation PAI-1 (coagulation) Ischémie
Abréviations
NE : Norépinéphrine ; ATS : Athérosclérose ; PAI : Inhibiteur de l’activité plasminogénique

Mise au point
Mise au point
22
En cas d’intolérance aux IEC, le candésartan a montré une réduction
de la morbimortalité cardiovasculaire chez les patients avec
insuffisance cardiaque de classe Il à Ill NYHA et dysfonction
systolique ventriculaire gauche. Le candésartan apporte
une amelioration du service médical rendu importante
(ASMR 2) dans la prise en charge de ces patients.
Avis de la commission de la transparence du 2 novembre 2005.
En association avec un IEC chez les patients restant
symptomatiques sous IEC, le candésartan a montré une
réduction de la morbimortalité cardiovasculaire en cas
d'insuffisance cardiaque de classe Il à Ill NYHA et
dysfonction systolique ventriculaire gauche. Chez
ces patients, le risque d'hyperkaliémie est augmenté. Le
candésartan apporte une amélioration du service médical
rendu modérée (ASMR 3) dans la prise en charge de ces patients.
Avis de la commission de la transparence du 2 novembre 2005.
INSUFFISANCE
CARDIAQUE
Classe II à III NYHA avec dysfonction systolique
ventriculaire gauche
(FEVG ≤40 %)
•
en cas d’intolérance aux IEC
•
ou en association avec un IEC chez les patients
restant symptomatiques sous IEC
DENOMINATION : KENZEN 32 mg comprimés sécables.FORME, PRESENTATION ET COMPOSITION : comprimés sécables dosés à 32 mg, de candesartan cilexétil (DCI). Boite de 28. Excipient à effet
notoire : lactose monohydraté. INDICATION THERAPEUTIQUE : Traitement de l’insuffisance cardiaque de classe II à III NYHA avec dysfonction systolique ventriculaire gauche (FEVG ≤40 %) : en cas d’intolérance
aux inhibiteurs de l’enzyme de conversion (IEC), ou en association avec un inhibiteur de l’enzyme de conversion (IEC) chez les patients restant symptomatiques sous IEC. POSOLOGIE ET MODE
D’ADMINISTRATION : la dose initiale habituellement recommandée est de 4 mg/jour en une prise. La posologie sera ensuite augmentée progressivement jusqu’à la dose cible de 32 mg/jour ou la plus
forte dose tolérée, en doublant la posologie à intervalles d’au moins 2 semaines. Sujet âgé, insuffisant rénal, diabétique : Risque majoré d’hyperkaliémie, potentiellement mortel. Traitements associés :
association possible aux autres traitements de l’IC : IEC, b-bloquants, diurétiques, digitaliques, ou avec une association de ces produits. La triple association de candesartan avec un IEC et un diurétique
hyperkaliémiant (ex. spironolactone ou éplérénone) est fortement déconseillée. Mode d’administration : une seule prise par jour pendant ou en dehors des repas. C.T.J : 1,37 €.CONTRE-
INDICATIONS :
Absolues :
hypersensibilité à l’un des constituants, à partir du 2ème trimestre de la grossesse, en cas de kaliémie > 5 mmol/L, de créatininémie > 265 micromol/l (> 30 mg/L) ou de
cl. créatinine < 30 ml/min.
Relatives :
diurétiques hyperkaliémiants (spironolactone, éplérénone, amiloride, triamtérène, seul ou associés…), potassium (sels de), sel de lithium ; au cours de
l’allaitement. MISES EN GARDE ET PRECAUTIONS PARTICULIERES D’EMPLOI : hypotension artérielle : la fréquence des hypotensions augmente avec l’âge, en cas de diabète ou d’association
à un autre traitement agissant sur le système rénine-angiotensine-aldostérone (SRAA) et peut nécessiter une réduction de la dose ou l’arrêt du traitement. Risque accru d’hypotension sévère et
d’insuffisance rénale en cas de sténose bilatérale de l’artère rénale ou de sténose artérielle rénale sur rein fonctionnel unique. Altération de la fonction rénale fréquemment observée. Sa fréquence
augmente avec l’âge, en cas de diabète ou d’association à un autre traitement agissant sur le SRAA et peut nécessiter une réduction de la dose ou l’arrêt du traitement. Aucune expérience
disponible chez les patients ayant eu une transplantation rénale récente. Augmenter progressivement les doses et surveiller la pression artérielle chez les patients hémodialysés. Risque
d’hyperkaliémie potentiellement mortel, majoré chez les sujets âgés, les insuffisants rénaux et les diabétiques, et/ou en cas d’association de plusieurs médicaments hyperkaliémiants [sels de
potassium, diurétiques hyperkaliémiants, IEC, ARA II, AINS (y compris inhibiteurs sélectifs de la COX 2), héparines (de bas poids moléculaires ou non fractionnées), immunosuppresseurs
comme la ciclosporine ou le tacrolimus, le triméthoprime], et/ou lors de la survenue d’évènements intercurrents [déshydratation, décompensation cardiaque aiguë, acidose métabolique,
altération de la fonction rénale, altération importante et soudaine de l’état général (ex. maladies infectieuses), souffrance et lyse cellulaire (ex. ischémie aiguë d’un membre, rhabdomyolyse,
traumatismes étendus). Avant d’associer plusieurs médicaments bloquant le SRAA, évaluer le rapport bénéfice/risque et considérer la possibilité d’une alternative thérapeutique. Le suivi
des patients insuffisants cardiaques devra comporter un ionogramme sanguin, avec contrôle de la kaliémie, de la natrémie, et de la fonction rénale avant instauration du traitement et
une semaine après ; et (avant et après) chaque augmentation de dose ou modification de traitement. En traitement d’entretien, réaliser les contrôles tous les mois pendant les trois
premiers mois, puis tous les trois mois la 1ère année et ensuite, tous les 6 mois ou lors de la survenue d’un événement intercurrent. KENZEN n’est pas indiqué dans l’insuffisance cardiaque
de classe IV NYHA. Risque d’hypotension au cours d’une anesthésie ou d’une intervention chirurgicale pouvant nécessiter le recours à un remplissage vasculaire et/ou à des substances
vasopressives. Prudence particulière chez les patients souffrant de sténose aortique ou mitrale, ou de cardiomyopathie obstructive hypertrophique. Utilisation non recommandée en
cas d’hyperaldostéronisme primaire. Risque d’hypotension aiguë, d’hyperazotémie, d’oligurie ou rarement d’insuffisance rénale aiguë chez les patients dont la tonicité vasculaire et la
fonction rénale dépendent de façon prédominante de l’activité du système rénine-angiotensine-aldostérone. Risque d’infarctus du myocarde ou d’accident vasculaire cérébral en cas
de cardiopathie ischémique ou d’affection cérébrovasculaire ischémique. Présence de lactose. Généralement déconseillé pendant le 1er trimestre de la grossesse. INTERACTIONS
AVEC D’AUTRES MEDICAMENTS ET AUTRES FORMES D’INTERACTIONS : Associations déconseillées : diurétiques hyperkaliémiants (spironolactone, éplérénone, amiloride,
triamtérène, seul ou associés…), potassium (sels de), Lithium (sels de). Associations nécessitant des précautions d’emploi : diurétiques, AINS. GROSSESSE ET
ALLAITEMENT : Grossesse : Par mesure de prudence, ne pas utiliser KENZEN au cours du 1er trimestre de la grossesse. L’administration de KENZEN est contre-indiquée
pendant les 2ème et 3ème trimestres de la grossesse. Allaitement : il est déconseillé d’administrer KENZEN. EFFETS SUR L’APTITUDE A CONDUIRE DES VEHICULES ET
A UTILISER DES MACHINES : Survenue occasionnelle de vertiges ou de fatigue. EFFETS INDESIRABLES : Observés le plus fréquemment (≥1/100, <1/10): altération
de la fonction rénale (augmentation de la créatininémie et/ou de l’urémie), hyperkaliémie, hypotension artérielle. Ces événements, potentiellement graves, et nécessitant
un suivi régulier au cours du traitement, étaient plus fréquents chez les patients > 70 ans, diabétiques, ou ayant reçu d’autres traitements agissant sur le SRAA.
SURDOSAGE : traitement
symptomatique et sur-
veiller les signes vitaux.
Le candesartan n’est pas
éliminé par hémodialyse.
PROPRIETES PHARMA-
CODYNAMIQUES : médica-
ment agissant sur le système
rénine-angiotensine/anta-
goniste de l’angiotensine II.
PROPRIETES PHARMA-
COCINETIQUES. DUREE
DE CONSERVATION : 2 ans.
PRECAUTIONS PARTICULIE-
RES DE CONSERVATION :
A conserver à une température ne
dépassant pas 25°C. PRESENTATION
ET NUMERO D’IDENTIFICATION
ADMINISTRATIVE : 28 comprimés
sous plaquettes thermoformées
(PVC/PVDC/Aluminium) : 368 707-8.
CONDITIONS DE PRESCRIPTION
ET DE DELIVRANCE : LISTE I. PRIX
PUBLIC TTC : 38,47 €. Spécialités
remboursées S.S à 65 % et agréées
Collec. TITULAIRE DE L’AUTORISA-
TION DE MISE SUR LE MARCHE :
Laboratoires TAKEDA, 11-15, quai
de Dion Bouton, 92816 PUTEAUX
cedex. Tél. : 01 46 25 16 16
- Fax : 01 46 97 00 11 - Info médic et
pharmacovigilance : 01 46 25 12 00.
Pour une information plus complète,
consulter le dictionnaire VIDAL.
DÉCOUVREUR DU CANDESARTAN
KE/F/119/01-06
candesartan cilexetil
®
nouveau dosage
32mg
annonce kenzen 210x270 1/06/06 16:35 Page 1
>>>
La Lettre du Cardiologue - n° 405 - mai 2007
EFFETS DÉLÉTÈRES DE L’ALDOSTÉRONE
Chez l’homme sain, un phénomène d’échappement à l’eff et de
l’aldostérone empêche qu’une administration continue d’aldosté-
rone ne produise une réabsorption sodique au-delà d’environ cinq
jours. Les mécanismes impliqués dans cet échappement semblent
multiples. Celui-ci résulterait de changements hémodynamiques
au niveau rénal, d’une up-regulation de la sécrétion de l’ANP et
d’une diminution des cotransporteurs Na
+
/Cl
-
thiazide-sensibles au
niveau des tubules rénaux distaux. En cas d’insuffi sance cardiaque
congestive, ce phénomène d’échappement est inopérant. En consé-
quence, même de petites quantités d’aldostérone vont provoquer
une rétention sodée et une expansion du volume. En outre, la
production cardiaque d’aldostérone est augmentée au cours de
l’insuffi sance cardiaque humaine, proportionnellement au niveau
de dysfonction ventriculaire (34). La production de l’enzyme de
conversion ainsi que celle du peptide natriurétique de type B sont
également activées dans les ventricules d’un cœur défaillant. Il
est donc probable que l’augmentation de la tension de paroi ou la
distension d’un ventricule dilaté observées lors d’une dysfonction
systolique soient des stimuli pour la production d’aldostérone,
d’enzyme de conversion et de peptide natriurétique de type B au
niveau des ventricules.
FIBROSE MYOCARDIQUE DUE À L’ALDOSTÉRONE
L’augmentation de l’aldostérone cardiaque est responsable d’une
fi brose myocardique et vasculaire. En eff et, l’aldostérone stimule
la synthèse du collagène par les fi broblastes cardiaques, induisant
une fi brose cardiaque, avec des conséquences défavorables sur
la fonction de pompe ainsi que sur le rythme cardiaque (eff et
arythmogène). Ce sont les travaux de Weber qui ont, les premiers,
montré que l’aldostérone associée au NaCl induisait une fi brose
cardiaque majeure chez le rat (35). Cette fi brose résulte d’un
eff et direct de l’aldostérone ; elle est indépendante des facteurs
hémodynamiques comme l’hypertension. L’eff et spécifi que de
l’aldostérone est confi rmé par les observations suivantes :
✓ le blocage de l’aldostérone par la spironolactone prévient la
fi brose, même à des doses qui n’infl uencent ni l’hypertension,
ni l’hypertrophie ventriculaire gauche ;
✓ la fi brose cardiaque induite par une infusion systémique d’al-
dostérone et de NaCl n’est pas prévenue par l’administration
intracérébroventriculaire de spironolactone, bien que ce traite-
ment prévienne l’apparition de l’hypertension artérielle.
Une étude récente (36) menée chez des patients normotendus
présentant un hyperaldostéronisme familial de type 1 démontre
clairement que c’est l’excès d’aldostérone en lui-même qui est
responsable des anomalies de structure et de fonction du ventri-
cule gauche. En eff et, ces patients atteints d’un hyperaldostéro-
nisme présentent, en l’absence d’hypertension, un remodelage
cardiaque avec épaississement du septum et de la paroi ventricu-
laire gauche, ainsi qu’une diminution de la fonction diastolique. La
fi brose cardiaque et le remodelage coronarien résultent donc bien
d’un eff et direct de l’aldostérone sur le tissu cardiaque. Cet eff et
trophique de l’aldostérone qui aboutit au remodelage vasculaire
est dépendant du sodium, ce qui suggère que l’aldostérone induit
une entrée de sodium dans les fi broblastes (35). Un travail récent
(37) a en outre démontré que, lors d’un infarctus, l’aldostérone
stimule l’activité des protéinases impliquées dans la production du
collagène et donc dans la fi brose cardiaque. En plus de l’aldosté-
rone et du sel, l’angiotensine II paraît également participer à cette
fi brose par l’intermédiaire des récepteurs AT1 (25). La synthèse
d’aldostérone cardiaque est augmentée dans la zone non infarcie
du cœur de rat un mois après la ligature coronaire, parallèlement
à la synthèse locale d’angiotensine II (27). Ainsi, l’aldostérone et
l’angiotensine II sont impliquées dans la régulation des processus
infl ammatoires et de réparation qui surviennent après lésion
tissulaire (38, 39). À cet eff et, elles stimulent la production des
cytokines et le chimiotactisme ; elles activent les macrophages aux
sites de réparation (40) et stimulent la croissance des fi broblastes
ainsi que la synthèse du collagène de type I et III qui gouvernent
la formation du tissu cicatriciel (37, 41). Le rôle physiologique de
la production locale d’aldostérone pourrait donc être son action
de réparation tissulaire après infarctus du myocarde (27).
EFFET ARYTHMOGÈNE DE L’ALDOSTÉRONE
Le rôle proarythmique de l’aldostérone (42) pourrait dépendre
partiellement de la fi brose myocardique, qui perturbe la fonction
électrique du cœur, de l’hypokaliémie et de l’hypomagnésémie
dues à la perte de potassium et de magnésium au niveau des
tubules rénaux, ainsi que de l’eff et de l’aldostérone sur la capture
myocardique des catécholamines (43, 44). Tous ces eff ets de
l’aldostérone prédisposent aux arythmies ventriculaires, à la
toxicité digitalique et donc à l’arrêt cardiaque (45).
ALDOSTÉRONE ET DYSFONCTION ENDOTHÉLIALE
L’aldostérone augmente la résistance périphérique et coro-
narienne en réponse à l’angiotensine II, à la sérotonine et à
la norépinéphrine. Elle induit la sécrétion d’endothéline, un
puissant stimulus de facteurs de croissance (46) qui favorise
le développement de l’athérosclérose. L’aldostérone inhibe en
outre la synthèse de NO des cellules vasculaires (47).
Finalement, l’aldostérone diminue le HDL-cholestérol, prédis-
posant ainsi à l’athérosclérose (48), et contribue, avec l’angio-
tensine II, à l’augmentation du potentiel de coagulation par
stimulation de la production du PAI-1 et de l’agrégation plaquet-
taire aux sites de saignement (49).
ANGIOTENSINE ET ALDOSTÉRONE :
FACTEURS DE RISQUE CARDIOVASCULAIRE
L’ensemble de ces eff ets délétères fait de l’aldostérone un facteur
de risque cardiovasculaire, indépendant de l’angiotensine II. De
nombreux travaux ont montré une relation entre la concentration

Mise au point
Mise au point
24
>>>
La Lettre du Cardiologue - n° 405 - mai 2007
plasmatique d’aldostérone et la mortalité chez les patients atteints
d’insuffi sance cardiaque, ainsi qu’avec la masse ventriculaire gauche,
qui est un bon indice de morbimortalité. Les inhibiteurs de l’en-
zyme de conversion (IEC) diminuent effi cacement la morbidité et la
mortalité des patients présentant une insuffi sance cardiaque et une
dysfonction ventriculaire gauche. Comme l’angiotensine II est un
stimulus important de la production d’aldostérone, il était raisonnable
et logique de penser que l’emploi d’un IEC aurait pour conséquence
de supprimer les eff ets indésirables à la fois de l’angiotensine II et de
l’aldostérone. Les résultats obtenus avec les IEC n’ont cependant pas
tout à fait répondu à l’attente, ce qui pourrait être dû à l’échappement
du blocage de la production d’aldostérone sous IEC. En eff et, des
observations cliniques ont mis en évidence un tel échappement,
qui se traduit par une remontée des concentrations plasmatiques
d’aldostérone après trois mois de traitement par IEC (43). Plusieurs
mécanismes ont été évoqués pour expliquer cet échappement. Il
pourrait s’agir d’un blocage incomplet du SRAA, d’une synthèse
d’angiotensine II empruntant d’autres voies que celle de l’ECA, d’une
stimulation de la sécrétion d’aldostérone en réponse à des infl uences
éventuellement combinées : hypokaliémie, hypomagnésémie chro-
nique, intervention de facteurs paracrines et neurocrines, et/ou
diminution du catabolisme hépatique de l’aldostérone (50).
L’angiotensine II, qu’elle provienne de la voie classique de l’ECA
ou de la voie alterne de la chymase, exerce diverses actions sur
le cœur. Par la stimulation de ses récepteurs AT1, elle agit au
niveau de la coagulation (profi brogènes) et pro-infl ammatoires.
L’infl ammation joue un rôle majeur dans la physiopathologie des
aff ections cardiaques. L’angiotensine II stimule en eff et l’expression
du facteur nucléaire κB (NF-κB), un facteur de transcription qui
règle l’expression génique des cytokines pro-infl ammatoires, telle
l’interleukine 6 (51). Au niveau cardiaque, l’angiotensine II joue
aussi un rôle important dans la prolifération vasculaire (52). Les
résultats d’études cliniques confi rment l’hypothèse selon laquelle
l’angiotensine II, produite indépendamment de la voie de l’ECA,
stimulerait la prolifération vasculaire et favoriserait ainsi la resté-
nose après une angioplastie coronarienne percutanée. En eff et,
la resténose est prévenue par l’administration d’un antagoniste
du récepteur de l’angiotensine II, alors qu’elle ne l’est pas en cas
d’administration d’un inhibiteur de l’ECA (53). Cela confi rme la
présence au niveau cardiaque ainsi que l’importance de la voie
alterne chymasique de la production de l’angiotensine II.
De nombreux modèles animaux de pathologie cardiaque (cardio-
myopathie, surcharge de pression, infarctus du myocarde) s’as-
socient à une élévation de la chymase cardiaque, aussi bien en ce
qui concerne l’ARNm que son activité enzymatique. Il en est de
même dans des homogénats de cœurs humains pathologiques
(cardiomyopathie idiopathique, infarctus du myocarde [54, 55]).
Toutes ces observations étayent l’hypothèse du rôle de l’angio-
tensine II, issue de la voie de la chymase, dans la pathogénie de
ces états pathologiques.
La chymase contribue aux aff ections cardiaques non seulement
par la transformation de l’angiotensine I en angiotensine II,
mais aussi par l’activation du TGF-β et par le remodelage de la
matrice extracellulaire. Ainsi, la chymase contribue à la fi brose
et à l’hypertrophie cardiaque.
EFFETS CARDIOVASCULAIRES BÉNÉFIQUES
RÉSULTANT DE L’INHIBITION DE LA CHYMASE
MASTOCYTAIRE
L’activité de la chymase cardiaque étant élevée dans de
nombreuses pathologies cardiaques, il était logique de penser
que son inhibition pourrait avoir un effet thérapeutique béné-
fique. C’est pourquoi plusieurs inhibiteurs sélectifs de cette
enzyme ont été développés ou sont en cours de développe-
ment (15). Ils n’ont pas encore été utilisés chez l’homme,
mais des résultats prometteurs ont été obtenus chez l’animal,
dans des modèles expérimentaux d’infarctus du myocarde,
de cardiomyopathie et d’insuffisance cardiaque induite par
tachycardie (15). Il est donc fort probable que ces inhibi-
teurs soient appelés à occuper une place significative dans
le traitement de maladies cardiaques où la dégranulation
des mastocytes, provoquée par la lésion ischémique, paraît
jouer un rôle important. Des inhibiteurs de la chymase ont
significativement réduit la prolifération vasculaire dans des
veines greffées chez le chien (56, 57), ainsi que la prolifération
vasculaire et l’hyperplasie intimale survenant après une lésion
par ballonnet de dilatation (58). Ces molécules pourraient
donc être utiles dans la prévention de la resténose après
dilatation percutanée de vaisseaux coronariens.
L’activité chymase est aussi augmentée dans la paroi des
anévrysmes de l’aorte abdominale (59), et son inhibition, dans
le modèle d’anévrysme du hamster, s’accompagne notamment
d’un eff et favorable sur le diamètre aortique, ce qui suggère que
l’inhibition de la chymase pourrait prévenir la progression de
l’anévrysme aortique abdominal (60).
L’inhibition de la chymase pourrait également avoir un eff et
thérapeutique dans certaines cardiomyopathies (15) [fi gure],
dans l’infarctus du myocarde (55), dans la prévention du remo-
delage cardiaque lors d’une insuffi sance cardiaque (61), ainsi
que dans l’athérosclérose (62).
Au niveau cardiaque, l’angiotensine II possède un effet
inotrope positif. Ainsi, la présence dans le cœur d’une voie
génératrice d’angiotensine II indépendante de l’ECA pourrait
avoir un rôle bénéfique important en cas de traitement de
l’insuffisance cardiaque par des IEC. Cette voie alterne n’étant
pas touchée par les IEC, l’angiotensine II qui en est issue
pourrait ainsi participer à la préservation de la fonction d’un
cœur insuffisant en maintenant son support inotrope positif.
En effet, en cas d’insuffisance cardiaque, les IEC améliorent
la fonction cardiaque par diminution des effets de l’angio-
tensine II au niveau des vaisseaux, réduisant ainsi les pré- et
post-charges cardiaques sans modifier l’effet inotrope positif
de l’angiotensine II cardiaque, puisque celle-ci provient de la
voie de la chymase, ce qui la rend indépendante de l’action
des IEC (10).
En conclusion, toutes les observations suggèrent que l’inhibition
de l’activité de la chymase par des inhibiteurs non peptidiques,
et donc actifs par voie orale, pourrait, dans un avenir proche,
faire partie de l’arsenal thérapeutique des pathologies cardio-
vasculaires.
 6
7
8
6
7
8
1
/
8
100%