Dossier thématique Aldostérone et cœur Aldosterone and the heart

143
Métabolismes Hormones Diabètes et Nutrition (X), n° 6, novembre-décembre 2006
Dossier
thématique
Aldostérone et cœur
Aldosterone and the heart
R.C. Gaillard*
L
e cœur est non seulement un
organe cible pour les actions
directes et indirectes de mul-
tiples hormones et facteurs hormo-
naux, mais il constitue également
un organe endocrine, au sens classi-
que du terme, puisqu’il synthétise et
sécrète dans la circulation des subs-
tances qui vont agir à distance.
Après la découverte du facteur
natriurétique auriculaire (ANF)
dans les années 1980, il a été montré
que le cœur fonctionnait comme un
véritable organe endocrine. En effet,
De Bold et al. (1) ont démontré en
1981 que l’injection à des animaux
de laboratoire d’extraits cardia-
ques d’origine auriculaire, mais
non d’origine ventriculaire, indui-
sait une natriurèse et une réduction
du volume intravasculaire. Cette
activité fut attribuée à l’ANF, qui,
après son isolement et sa carac-
térisation, est devenu le peptide
natriurétique auriculaire (ANP).
Celui-ci est synthétisé par le cœur,
d’où il est sécrété dans la circulation
pour agir au niveau rénal, où il se
comporte comme un puissant agent
natriurétique, ainsi qu’au niveau
vasculaire, où il induit une vasodi-
latation. L’ANP agit également sur
la contractilité du myocarde. Depuis
la découverte de l’ANP, d’autres
peptides ou facteurs humoraux
cardiaques ont été identifiés. Parmi
eux, citons l’adrénomédulline, dont
la synthèse est stimulée par une
surcharge de pression et de volume
(2), et l’urocortine, un nouveau
membre des peptides de la famille
du CRH (3). Ces deux peptides sont
de puissants vasodilatateurs. En
outre, l’adrénomédulline diminue
le remodelage vasculaire par un
effet antiprolifératif au niveau de
la paroi vasculaire, et pourrait ainsi
exercer une protection contre le
développement de l’athérosclérose
et/ou de la resténose (4, 5). Le cœur
est également capable, comme nous
le mentionnerons dans cette brève
revue, de produire lui-même tous
les composants du système rénine-
angiotensine-aldostérone (SRAA).
Il produit encore de nombreux
autres facteurs humoraux tels que
le monoxyde d’azote (NO), l’en-
dothéline, les éicosanoïdes et les
substances du système kallicréine-
kinine. Toutes ces substances sont
des modulateurs de la performance
cardiaque, que ce soit dans des
circonstances physiologiques ou
physiopathologiques (6).
Le cœur constitue aussi un organe
cible pour des hormones et certains
facteurs humoraux, comme les
catécholamines, les substances du
SRAA, les estrogènes, la testos-
* Service d’endocrinologie, diabétologie et méta-
bolisme, CHU Vaudois, 1011 Lausanne, Suisse.
Mots-clés : Système rénine-angiotensine-aldostérone – Chymase
mastocytaire – Fibrose myocardique – Arythmie – Récepteur miné-
ralocorticoïde – Spironolactone – Éplérénone.
Keywords: Renin angiotensin aldosterone system – Mast cells chy-
mase – Myocardial fibrosis – Arrhythmia – Mineralcorticoid recep-
tor – Spironolactone – Eplerone.
Résumé
L
e cœur est non seulement un organe cible pour les actions hor-
monales mais aussi un organe endocrine qui synthétise et sécrète
des hormones. Ainsi le cœur est-il à la fois un organe cible de
l’aldostérone et de l’angiotensine II, dont il est aussi une source. Il pro-
duit de l’angiotensine II soit par la voie classique du système rénine-
angiotensine-aldostérone, dépendante de l’enzyme de conversion de
l’angiotensine (ECA), soit par une voie alterne indépendante de cette
dernière, la voie de la chymase (une sérine protéinase présente dans
les mastocytes). Outre ses effets classiques qui assurent l’homéostasie
hydro-électrolytique, l’aldostérone induit de nombreux effets délétè-
res (fibrose myocardique, dysfonction endothéliale, augmentation du
PAI-1, etc.) responsables d’une augmentation de la morbidité et de
la mortalité cardiovasculaires. Associé au traitement conventionnel,
le blocage spécifique du récepteur minéralocorticoïde par de faibles
doses de spironolactone ou d’éplérénone réduit de manière très signi-
ficative cette morbi-mortalité chez des patients atteints d’une insuffi-
sance cardiaque modérée à sévère et/ou d’une dysfonction ventricu-
laire gauche après infarctus du myocarde.

144
Métabolismes Hormones Diabètes et Nutrition (X), n° 6, novembre-décembre 2006
thématique
Dossier
térone, l’insuline, ainsi que pour
l’hormone de croissance et les
sécrétines peptidiques de l’hor-
mone de croissance (6-9). Ainsi,
dans l’insuffisance cardiaque, le
cœur est incapable d’assurer correc-
tement sa fonction de pompe, et la
baisse du débit cardiaque qui en
résulte entraîne une augmentation
des concentrations plasmatiques
de plusieurs substances actives au
niveau cardiovasculaire, telles les
catécholamines, l’angiotensine II,
l’aldostérone, la vasopressine et
l’endothéline. L’action de toutes
ces substances a pour but de main-
tenir la pression de perfusion péri-
phérique. À long terme, toutefois,
les actions de ces hormones se
révèlent dommageables, car elles
sont responsables d’effets délé-
tères sur la structure et la fonction
cardiaques, et contribuent ainsi
paradoxalement à l’aggravation
de la dysfonction cardiaque. L’ac-
tivation de ces systèmes neuro-
hormonaux sert de marqueur de
l’insuffisance cardiaque et constitue
ainsi une cible thérapeutique. Parmi
ces hormones, il a été récemment
démontré que les composants du
SRAA ont effectivement des effets
spécifiques et néfastes sur le cœur
et les vaisseaux, et que le blocage
de leur action pourrait ainsi être
bénéfique.
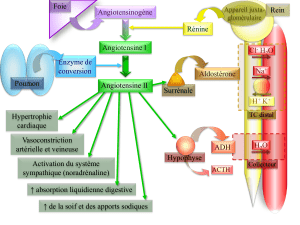
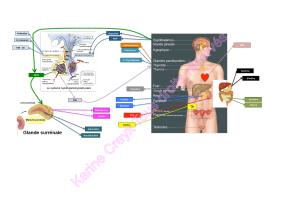
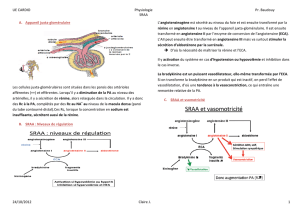
Le système rénine-
angiotensine-aldostérone
Le SRAA préserve l’homéostasie
circulatoire lors d’une perte de sel
et d’eau résultant par exemple d’une
transpiration intense et prolongée,
de vomissements ou d’une diarrhée.
La rénine, l’angiotensine II et l’al-
dostérone sont des éléments clés de
ce système. La rénine, synthétisée
par l’appareil juxtaglomérulaire du
rein, produira le clivage de quatre
acides de l’angiotensinogène circu-
lant (le précurseur hépatique de
tous les peptides de l’angiotensine)
pour former l’angiotensine I, un
décapeptide biologiquement inactif.
L’enzyme de conversion de l’an-
giotensine (ECA) qui est liée à la
membrane des cellules endothéliales
effectue ensuite le clivage de deux
acides aminés de l’angiotensine I
pour engendrer l’angiotensine II.
La chymase mastocytaire
L’ECA est donc une enzyme indis-
pensable à la génération de l’angio-
tensine II. Dans le cœur, il existe
cependant une voie alterne ne
passant pas par l’ECA, mais impli-
quant l’α-chymase, une enzyme
similaire à la chymotrypsine qui est
exprimée dans les granules sécré-
toires des mastocytes cardiaques.
La présence d’une telle voie a été
démontrée in vitro, puisque, dans
des homogénats de tissu cardiaque
humain, 75 % de l’angiotensi-
ne II formée sont dépendants de
la chymase, alors que seuls 25 %
le sont de la voie classique de
l’ECA (10, 11). Diverses études
ont confirmé que l’angiotensine II,
dans les divers tissus d’origine
cardiovasculaire, est dépendante de
la voie de la chymase plutôt que de
la voie classique de l’ECA, puisque
la production d’angiotensine II dans
ces tissus est bloquée à près de 90 %
par des inhibiteurs de la chymase
(12-14).
La chymase est stockée dans les
granules des mastocytes sous forme
inactive, en raison d’un pH à 5,5
dans ces granules. Le pH optimal
pour l’activité enzymatique est
compris entre 7 et 9, situation
rencontrée lors de la sécrétion de
la chymase après activation des
mastocytes présents dans un tissu
lésé ou inflammatoire (15). L’acti-
vité chymasique ne s’exerce qu’au
niveau tissulaire local, car de puis-
sants inhibiteurs de la chymase sont
présents dans la circulation sanguine
et bloquent immédiatement son acti-
vité enzymatique (15).
Il existe deux types de chymase, la
forme α, qui prédomine notamment
chez l’homme, le singe et le mouton,
et la forme β, que l’on trouve chez
le rat et la souris (16). La chymase
cardiaque humaine présente une
très grande spécificité pour l’an-
giotensine I, ce qui la distingue
d’autres enzymes impliquées dans
la production de l’angiotensine II,
telles l’ECA, la kallicréine, la
cathepsine et les autres chymases
(10). En outre, la chymase cardiaque
humaine, au contraire de l’ECA, ne
dégrade pas l’angiotensine II ni les
autres peptides comme la bradyki-
nine, la substance P et la gonadoli-
bérine (10).
La chymase mastocytaire contribue
non seulement à la transformation de
l’angiotensine I en angiotensine II,
mais elle participe aussi à la sécré-
tion et à l’activation de la cytokine
IL-1β, une cytokine pro-inflamma-
toire, et du TGF-β1, une cytokine
qui est impliquée notamment dans
l’hypertrophie et la fibrose cardiaque
(17, 18). La chymase active le TGF-
β qui se trouve sous forme inactive
ou latente dans les mastocytes (18).
Les mastocytes sont donc, par ce
mécanisme, impliqués dans des
processus pathologiques caracté-
risés par des événements inflamma-
toires et fibrogéniques tels que la
fibrose pulmonaire (19), l’infarctus
du myocarde et la fibrose myocar-
dique post-transplantation (20, 21).
Toutes ces observations suggèrent
qu’une inhibition de ces effets par
des inhibiteurs de la chymase pour-
rait être utilisée à but thérapeutique
pour prévenir les maladies cardio-
vasculaires et la fibrose.
En plus des effets susmentionnés,
l’α-chymase est aussi capable de
former les endothélines de 31 acides
aminés, de dégrader l’endothéline 1,
de modifier le métabolisme lipi-
dique et d’endommager la matrice
extracellulaire (22).
L’angiotensine II exerce de
nombreuses actions pour maintenir
l’homéostasie circulatoire, indui-
sant la constriction des artérioles
au niveau des circulations rénale
et systémique, ainsi que la réab-
sorption du sodium au niveau des

145
Métabolismes Hormones Diabètes et Nutrition (X), n° 6, novembre-décembre 2006
Dossier
thématique
segments proximaux du néphron.
L’angiotensine II stimule égale-
ment la sécrétion d’aldostérone
responsable d’une réabsorption du
sodium (en échange du potassium)
au niveau des segments distaux du
néphron, ainsi que dans le côlon,
les glandes salivaires et sudoripares.
Lorsque le volume intravasculaire
est réduit, l’angiotensine II est le
stimulus principal de la production
d’aldostérone, qui peut cependant
aussi être stimulée par le potassium,
l’ACTH, les catécholamines, l’en-
dothéline et la sérotonine d’origine
mastocytaire. Parmi ces stimuli de
l’aldostérone, le potassium a un rôle
physiologique important, puisqu’il
permet de maintenir son homéo-
stasie grâce à la capacité de l’aldo-
stérone, dont il stimule la sécrétion,
à augmenter l’excrétion potassique
dans les urines, les selles, la sueur et
la salive (23). L’aldostérone permet
ainsi de prévenir l’hyperkaliémie en
cas d’apport potassique élevé dans
l’alimentation ou après un exercice
physique intense qui entraîne une
sortie de potassium des muscles.
L’aldostérone produit deux types
d’action : des modifications immé-
diates de conductances ioniques,
ainsi que l’induction différée de
gènes cibles qui survient après
liaison sur son récepteur intracellu-
laire, le récepteur des minéralocorti-
coïdes (MR). Son action principale
consiste à augmenter la synthèse
et l’activité des canaux sodiques
amiloride-sensibles et la Na/K-
ATPase membranaire des cellules
du tubule contourné distal et du
tube collecteur rénal pour aboutir à
la réabsorption du sodium. Comme
nous le verrons ci-après, l’aldosté-
rone, parallèlement à cette action
essentielle de réabsorption sodique,
exerce d’autres actions, notamment
sur le système cardiovasculaire.
Le récepteur MR de l’aldostérone est
un facteur de transcription, membre
de la superfamille des récepteurs
nucléaires, qui comprend les récep-
teurs aux hormones stéroïdes/
thyroïdes et à l’acide rétinoïque. Ces
récepteurs sont classiquement loca-
lisés dans des cellules épithéliales
(côlon, reins, glandes salivaires et
sudoripares). Des études récentes
ont toutefois démontré que les MR
pouvaient également être présents
dans des tissus non épithéliaux tels
que les organes circumventriculaires
du cerveau, le cœur et les vaisseaux
sanguins. Les domaines de fixation
sur l’ADN et de liaison au ligand
du MR et du récepteur des gluco-
corticoïdes (GR) ont une grande
homologie de séquences. Pour cette
raison, le MR peut également lier le
cortisol avec la même affinité que
pour l’aldostérone. Le cortisol peut
être un véritable concurrent pour
l’aldostérone, car sa concentration
plasmatique est 100 à 1 000 fois
supérieure à celle de l’aldostérone.
Aussi, pour “protéger” le MR des
glucocorticoïdes, les cellules dispo-
sent-elles d’une enzyme, la 11-β-
hydroxystéroïde deshydrogénase
(11-β-HSD) de type 2, qui trans-
forme le cortisol (actif) en corti-
sone, inactive car incapable de se
lier au MR. Cela suppose donc que,
dans les types cellulaires spécifi-
quement répondeurs à l’aldosté-
rone, il y ait une colocalisation de
la 11-β-HSD 2 et du MR. Une telle
colocalisation a été vérifiée dans le
rein, et elle est aussi présente dans
le cœur humain (24).
Synthèse de l’aldostérone
La synthèse de l’aldostérone a lieu
dans le cortex surrénalien, où la
P-450 aldostérone-synthase, sous
contrôle de l’angiotensine II et du
potassium, et plus faiblement de
l’ACTH et du sodium, catalyse
cette synthèse à partir de la désoxy-
corticostérone. Des sites de produc-
tion extrasurrénaliens ont cependant
été identifiés. Il s’agit du cœur, de
l’aorte, de l’artère pulmonaire et
du cerveau. Un SRAA complet a
été mis en évidence au niveau du
cœur (25), qui possède ainsi l’en-
semble de l’équipement enzyma-
tique nécessaire à la production
d’aldostérone (26). Le cœur est
également un organe cible pour
les effets de l’aldostérone, puisque
le messager du MR est abondant
dans les cardiomyocytes et la paroi
endothéliale des artères principales
(24). La concentration tissulaire
cardiaque d’aldostérone, qui est
déjà 15 fois supérieure à celle du
plasma, peut encore augmenter de
deux à trois fois dans le modèle
murin de situation postinfarctus par
ligature coronaire (27). Toutefois,
la contribution cardiaque aux taux
plasmatiques d’aldostérone paraît
non significative comparativement
à celle du cortex surrénalien (28).
Si elle ne contribue pas aux taux
circulants de cette hormone, elle
joue certainement un rôle paracrine
et autocrine important au niveau
cardiaque. Il est intéressant de noter
que le sel a une fonction régulatrice
différente sur l’aldostérone plasma-
tique et sur l’aldostérone cardiaque.
En effet, un régime hypersodé chez
le rat normotendu induit une dimi-
nution de l’aldostérone plasma-
tique, alors qu’une augmentation
de l’aldostérone cardiaque et une
hypertrophie du cœur sont relevées
(29). Cette observation suggère
donc qu’un régime hypersodé
augmente la synthèse d’aldostérone
du cœur et contribue ainsi à l’hy-
pertrophie cardiaque de manière
indépendante du SRAA circulant.
Des travaux récents ont clairement
démontré que l’aldostérone exerçait
des effets directs sur le cardiomyo-
cyte, par l’induction, en présence
d’une concentration extracellulaire
de sodium, d’une hypertrophie
cardiaque (30). De plus, au niveau
du cœur, l’aldostérone exerce un
rétrocontrôle positif sur sa propre
production, puisqu’elle augmente
paradoxalement l’expression de
l’ARN messager de l’enzyme de
conversion. Ce mécanisme pourrait
être responsable de l’augmentation
et de la progression de l’activité du
SRAA cardiaque en cas d’insuffi-
sance cardiaque.
Le cœur est donc non seulement un
organe cible pour les minéralocorti-

146
Métabolismes Hormones Diabètes et Nutrition (X), n° 6, novembre-décembre 2006
thématique
Dossier
Tableau. Conséquences délétères de l’augmentation de l’aldostérone.
Rétention sodée Insuffisance cardiaque
Perte potassique Arythmies ventriculaires
Perte de magnésium Toxicité digitalique
Fibrose myocardique
Production de collagène du myocarde Dysfonction ventriculaire
Hypertrophie ventriculaire
Diminution de la réserve coronarienne Ischémie
Dysfonction des barorécepteurs Hypertension artérielle
Diminution capture myocardique NE Arythmies
Dysfonction endothéliale ATS
Diminution du HDL-cholestérol ATS
Augmentation PAI-1 (coagulation) Ischémie
Abréviations
NE : Norépinéphrine ; ATS : Athérosclérose ; PAI : Inhibiteur de l’activité plasminogénique
coïdes et les glucocorticoïdes, mais
il semble également constituer un
organe endocrine capable de synthé-
tiser de novo des corticostéroïdes. Il
y a cependant controverse quant à
l’origine précise de l’aldostérone
cardiaque (31-33), des arguments
scientifiques solides amenant à
considérer que l’aldostérone intra-
cardiaque provient essentiellement
de la circulation générale (28).
Effets de l’aldostérone
Les effets minéralocorticoïdes classi-
ques incluent la rétention du sodium
et de l’eau ainsi que l’excrétion du
potassium et du magnésium. Ils
contrôlent l’homéostasie du sel et
de l’eau. Depuis quelques années,
de nouveaux effets ont été mis en
évidence, en particulier au niveau du
cœur et des vaisseaux. Ces derniers
sont plutôt considérés comme des
effets indésirables. Ils sont respon-
sables de l’induction d’une fibrose
cardiaque importante, d’une fibrose/
rigidité vasculaire, d’une diminution
de la capture myocardique de norépi-
néphrine, d’une augmentation de la
production de l’inhibiteur de l’activa-
teur du plasminogène de type 1 (PAI-
1), de l’induction d’une dysfonction
endothéliale, et d’une diminution du
HDL-cholestérol. Autant d’effets
dont les conséquences délétères sont
résumées dans le tableau.
Effets délétères
de l’aldostérone
Chez l’homme sain, un phénomène
d’échappement à l’effet de l’aldosté-
rone empêche qu’une administration
continue d’aldostérone ne produise
une réabsorption sodique au-delà
d’environ cinq jours. Les méca-
nismes impliqués dans cet échappe-
ment semblent multiples. Celui-ci
résulterait de changements hémody-
namiques au niveau rénal, d’une up-
regulation de la sécrétion de l’ANP
et d’une diminution des cotranspor-
teurs Na+/Cl- thiazide-sensibles au
niveau des tubules rénaux distaux. En
cas d’insuffisance cardiaque conges-
tive, ce phénomène d’échappement
est inopérant. En conséquence,
même de petites quantités d’aldos-
térone vont provoquer une rétention
sodée et une expansion du volume.
En outre, la production cardiaque
d’aldostérone est augmentée au
cours de l’insuffisance cardiaque
humaine, proportionnellement au
niveau de dysfonction ventriculaire
(34). La production de l’enzyme de
conversion ainsi que celle du peptide
natriurétique de type B sont égale-
ment activées dans les ventricules
d’un cœur défaillant. Il est donc
probable que l’augmentation de la
tension de paroi ou la distension
d’un ventricule dilaté observées lors
d’une dysfonction systolique soient
des stimuli pour la production d’al-
dostérone, d’enzyme de conversion
et de peptide natriurétique de type B
au niveau des ventricules.
Fibrose myocardique due
à l’aldostérone
L’augmentation de l’aldostérone
cardiaque est responsable d’une
fibrose myocardique et vasculaire.
En effet, l’aldostérone stimule la
synthèse du collagène par les fibro-
blastes cardiaques, induisant une
fibrose cardiaque, avec des consé-
quences défavorables sur la fonction
de pompe ainsi que sur le rythme
cardiaque (effet arythmogène). Ce
sont les travaux de Weber qui ont,
les premiers, montré que l’aldosté-
rone associée au NaCl induisait une
fibrose cardiaque majeure chez le
rat (35). Cette fibrose résulte d’un
effet direct de l’aldostérone ; elle
est indépendante des facteurs hémo-
dynamiques comme l’hypertension.
L’effet spécifique de l’aldostérone
est confirmé par les observations
suivantes :
le blocage de l’aldostérone par la
spironolactone prévient la fibrose,
même à des doses qui n’influencent
ni l’hypertension, ni l’hypertrophie
ventriculaire gauche ;
la fibrose cardiaque induite par
une infusion systémique d’aldosté-
rone et de NaCl n’est pas prévenue
par l’administration intracérébro-
ventriculaire de spironolactone, bien
que ce traitement prévienne l’appa-
rition de l’hypertension artérielle.
✓
✓

147
Métabolismes Hormones Diabètes et Nutrition (X), n° 6, novembre-décembre 2006
Dossier
thématique
Une étude récente (36) menée chez
des patients normotendus présentant
un hyperaldostéronisme familial de
type 1 démontre clairement que c’est
l’excès d’aldostérone en lui-même
qui est responsable des anomalies
de structure et de fonction du ventri-
cule gauche. En effet, ces patients
atteints d’un hyperaldostéronisme
présentent, en l’absence d’hyper-
tension, un remodelage cardiaque
avec épaississement du septum et de
la paroi ventriculaire gauche, ainsi
qu’une diminution de la fonction
diastolique. La fibrose cardiaque et
le remodelage coronarien résultent
donc bien d’un effet direct de l’al-
dostérone sur le tissu cardiaque. Cet
effet trophique de l’aldostérone qui
aboutit au remodelage vasculaire
est dépendant du sodium, ce qui
suggère que l’aldostérone induit
une entrée de sodium dans les fibro-
blastes (35). Un travail récent (37)
a en outre démontré que, lors d’un
infarctus, l’aldostérone stimule l’ac-
tivité des protéinases impliquées
dans la production du collagène
et donc dans la fibrose cardiaque.
En plus de l’aldostérone et du sel,
l’angiotensine II paraît également
participer à cette fibrose par l’inter-
médiaire des récepteurs AT1 (25).
La synthèse d’aldostérone cardiaque
est augmentée dans la zone non
infarcie du cœur de rat un mois
après la ligature coronaire, parallè-
lement à la synthèse locale d’angio-
tensine II (27). Ainsi, l’aldostérone
et l’angiotensine II sont impliquées
dans la régulation des processus
inflammatoires et de réparation qui
surviennent après lésion tissulaire
(38, 39). À cet effet, elles stimulent
la production des cytokines et le
chimiotactisme ; elles activent les
macrophages aux sites de réparation
(40) et stimulent la croissance des
fibroblastes ainsi que la synthèse du
collagène de type I et III qui gouver-
nent la formation du tissu cicatriciel
(37, 41). Le rôle physiologique de
la production locale d’aldostérone
pourrait donc être son action de
réparation tissulaire après infarctus
du myocarde (27).
Effet arythmogène
de l’aldostérone
Le rôle proarythmique de l’al-
dostérone (42) pourrait dépendre
partiellement de la fibrose
myocardique, qui perturbe la
fonction électrique du cœur, de
l’hypokaliémie et de l’hypo-
magnésémie dues à la perte de
potassium et de magnésium au
niveau des tubules rénaux, ainsi
que de l’effet de l’aldostérone sur
la capture myocardique des caté-
cholamines (43, 44). Tous ces
effets de l’aldostérone prédispo-
sent aux arythmies ventriculaires,
à la toxicité digitalique et donc à
l’arrêt cardiaque (45).
Aldostérone
et dysfonction endothéliale
L’aldostérone augmente la résistance
périphérique et coronarienne en
réponse à l’angiotensine II, à la séro-
tonine et à la norépinéphrine. Elle
induit la sécrétion d’endothéline,
un puissant stimulus de facteurs de
croissance (46) qui favorise le déve-
loppement de l’athérosclérose. L’al-
dostérone inhibe en outre la synthèse
de NO des cellules vasculaires (47).
Finalement, l’aldostérone diminue
le HDL-cholestérol, prédisposant
ainsi à l’athérosclérose (48), et
contribue, avec l’angiotensine II,
à l’augmentation du potentiel de
coagulation par stimulation de la
production du PAI-1 et de l’agré-
gation plaquettaire aux sites de
saignement (49).
Angiotensine
et aldostérone : facteurs
de risque cardiovasculaire
L’ensemble de ces effets délétères
fait de l’aldostérone un facteur
de risque cardiovasculaire, indé-
pendant de l’angiotensine II. De
nombreux travaux ont montré une
relation entre la concentration
plasmatique d’aldostérone et la
mortalité chez les patients atteints
d’insuffisance cardiaque, ainsi
qu’avec la masse ventriculaire
gauche, qui est un bon indice de
morbimortalité. Les inhibiteurs de
l’enzyme de conversion (IEC) dimi-
nuent efficacement la morbidité et
la mortalité des patients présentant
une insuffisance cardiaque et une
dysfonction ventriculaire gauche.
Comme l’angiotensine II est un
stimulus important de la production
d’aldostérone, il était raisonnable
et logique de penser que l’emploi
d’un IEC aurait pour conséquence
de supprimer les effets indésirables
à la fois de l’angiotensine II et de
l’aldostérone. Les résultats obtenus
avec les IEC n’ont cependant pas
tout à fait répondu à l’attente, ce
qui pourrait être dû à l’échappe-
ment du blocage de la production
d’aldostérone sous IEC. En effet,
des observations cliniques ont mis
en évidence un tel échappement,
qui se traduit par une remontée
des concentrations plasmatiques
d’aldostérone après trois mois de
traitement par IEC (43). Plusieurs
mécanismes ont été évoqués pour
expliquer cet échappement. Il pour-
rait s’agir d’un blocage incomplet
du SRAA, d’une synthèse d’an-
giotensine II empruntant d’autres
voies que celle de l’ECA, d’une
stimulation de la sécrétion d’aldos-
térone en réponse à des influences
éventuellement combinées : hypo-
kaliémie, hypomagnésémie chro-
nique, intervention de facteurs
paracrines et neurocrines, et/ou
diminution du catabolisme hépa-
tique de l’aldostérone (50).
L’angiotensine II, qu’elle
provienne de la voie classique de
l’ECA ou de la voie alterne de la
chymase, exerce diverses actions
sur le cœur. Par la stimulation de
ses récepteurs AT1, elle agit au
niveau de la coagulation (profi-
brogènes) et pro-inflammatoires.
 6
7
8
9
10
6
7
8
9
10
1
/
10
100%