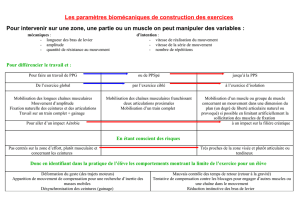
Myopathies des ceintures - site de l`association GENS

Myopathies des ceintures
[17-175-C-10] - Doi : 10.1016/S0246-0378(10)53745-X
G. Solé : Praticien hospitalier
Introduction
Le terme de myopathie des ceintures est longtemps resté décrié, en raison de l'hétérogénéité
des pathologies que l'on peut classer sous cette dénomination. Certains spécialistes ont voulu
abandonner ce concept il y a quelques années. On lui préfère actuellement le terme plus
restrictif de dystrophie des ceintures. Ces myopathies ont en commun la présence d'un déficit
musculaire des ceintures associé à des lésions histologiques dites « dystrophiques », c'est-à-
dire associant nécrose et régénération. On distingue les formes autosomiques dominantes, au
nombre de sept actuellement, des formes récessives, au nombre de 15. Devant la découverte
permanente de nouveaux gènes, il est fondamental de rappeler l'importance des aspects
cliniques qui peuvent orienter les explorations anatomopathologiques et génétiques. Après un
bref rappel historique, nous étudions d'abord les éléments communs à toutes les dystrophies
des ceintures, puis les caractéristiques cliniques et paracliniques propres à chacune de ces
pathologies. Dans un troisième temps, nous proposons une stratégie de diagnostic étiologique.
Historique
De la description clinique aux aspects biopsiques
Les premières descriptions de myopathie des ceintures remontent à la fin du XIXe siècle.
Leyden fut le premier à décrire, en 1876, une forme de dystrophie musculaire plus bénigne
que la myopathie de Duchenne [1]. Dans la première moitié du XXe siècle, plusieurs tentatives
de classification se sont succédées et le concept de myopathie des ceintures a été fortement
débattu [2]. Après la Seconde Guerre mondiale, le diagnostic des pathologies
neuromusculaires connaît un nouvel essor grâce au développement des techniques
d'électrophysiologie et surtout de microscopie optique puis électronique. C'est Stevenson qui
introduisit, en 1953, le terme de dystrophie des ceintures autosomiques (autosomal limb-
girdle muscular dystrophy [LGMD]) à partir d'une série de 51 familles irlandaises [3]. La
classique série de Walton et Nattrass fut la première à individualiser clairement les
myopathies des ceintures autosomiques récessives [4]. En raison de l'hétérogénéité clinique et
des progrès des techniques histologiques, la nécessité de conserver ce terme de dystrophie des
ceintures a été longuement discutée. Dans les années 1980, le terme plus général de «
syndrome des ceintures » était alors communément utilisé [5].
Ère de la biologie moléculaire : définition et classification actuelle
Les progrès de la biologie moléculaire ont permis d'individualiser les différentes dystrophies
des ceintures et de clarifier en partie leur hétérogénéité. Ces progrès moléculaires ont rendu
nécessaire l'établissement d'une classification. En 1995, une définition de travail [6] et une
classification des dystrophies des ceintures [7] ont été proposées. Les dystrophies des
ceintures se définissent par un déficit moteur proximal, épargnant les muscles distaux, faciaux
et oculomoteurs à la phase précoce de la pathologie et pour lequel la biopsie individualise des

lésions dystrophiques. La classification repose sur le mode de transmission : LGMD1 pour les
formes autosomiques dominantes et LGMD2 pour les formes autosomiques récessives.
Chaque pathologie est ensuite désignée par une lettre unique, attribuée principalement selon
l'ordre chronologique des publications. Le Tableau 1 récapitule la classification actualisée en
2007 [8]. Les principales protéines impliquées dans les dystrophies musculaires des ceintures
sont schématisées dans la Figure 1.
L'orientation diagnostique entre ces diverses formes se fait à partir d'un faisceau d'arguments :
hérédité, présentation clinique, données anatomopathologiques, étude des protéines
musculaires sur lame et en western blot.
Caractéristiques communes
Toutes les dystrophies des ceintures présentent des aspects cliniques et paracliniques
communs.
Aspects cliniques
L'âge de début est variable en fonction du type de dystrophie des ceintures. Les troubles
débutent généralement à la ceinture pelvienne par une marche dandinante, éventuellement
associée à des chutes. Les patients ont des difficultés à monter des escaliers et doivent s'aider
de la rampe. Le relevé est myopathique avec un signe de Gowers. L'aggravation est presque
toujours lentement progressive. La perte de la marche est inconstante. L'atteinte de la ceinture
scapulaire se manifeste initialement par une difficulté à porter des charges lourdes. La
musculature axiale peut être touchée dans certaines formes. Si cette atteinte débute avant la
fin de la croissance, elle peut être responsable d'une scoliose. L'étude de la force musculaire
analytique confirme le déficit prédominant aux ceintures et permet de rechercher une atteinte
d'autres muscles (comme les muscles distaux) qui peut orienter vers une étiologie spécifique.
On recherche aussi une pseudohypertrophie des mollets ou de la langue du type de celle
observée dans les maladies de Duchenne et de Becker. Comme dans la majorité des atteintes
musculaires primitives, on ne retrouve pas de déficit sensitif ni d'atteinte centrale, sauf
exceptions. Il est fondamental de rechercher des signes associés comme un déficit distal, une
atteinte de la musculature bulbaire, des rétractions ostéotendineuses ou des troubles cognitifs.
Aspects paracliniques
Trois examens principaux peuvent orienter le diagnostic. Le dosage des créatine-kinases (CK)
est couramment réalisé. Leur élévation témoigne d'une lésion évolutive de la fibre musculaire.
Elles sont fréquemment plus élevées en début de maladie. Le taux peut se normaliser par la
suite en raison de la dégénérescence fibroadipeuse du tissu musculaire. L'imagerie musculaire
est aussi souvent utile. Le scanner et l'imagerie par résonance magnétique (IRM) ont un
apport généralement identique dans le cadre des dystrophies. On recherche la présence d'une
amyotrophie et/ou d'une dégénérescence graisseuse et on analyse sa topographie. En scanner,
le muscle normal présente une densité homogène, alors que le muscle pathologique apparaît
hypodense et hétérogène (souvent marbré). Les aspects IRM sont globalement proches et
dépendent des séquences utilisées. Le scanner est plus volontiers utilisé en raison de sa
rapidité d'exécution et de sa meilleure disponibilité. L'IRM est en revanche bien plus utile s'il
existe une hésitation entre myopathie héréditaire et myopathie inflammatoire. La topographie
de l'atteinte peut orienter le diagnostic étiologique. L'électromyogramme (EMG) a une place

plus limitée. Il confirme généralement l'existence de signes myogènes, mais peut parfois être
pris en défaut. Il est utile pour identifier certains diagnostics différentiels : dystrophie
myotonique de type 2, atteinte neurogène, etc.
Aspects anatomopathologiques
La biopsie musculaire reste l'examen de choix afin de parvenir au diagnostic étiologique. Par
opposition aux myopathies congénitales où il existe une anomalie du développement de la
fibre musculaire, les dystrophies des ceintures se caractérisent par l'existence d'une nécrose
des fibres matures. Afin de maintenir la fonction tissulaire, cette nécrose est suivie par une
régénération. Ces deux phénomènes se succèdent en permanence et leur association sur la
biopsie définit la lésion dystrophique [9].
Nécrose
Quand une lésion se produit sur la membrane de la fibre musculaire (sarcolemme), elle
entraîne une entrée de calcium conduisant à une contraction anormalement soutenue des
myofibrilles (unité contractile des fibres musculaires) puis à leur destruction. La région lésée
est détruite par des protéases intrinsèques à la fibre musculaire. Les noyaux subissent alors
une caryolyse complète. Sur le plan histologique, ces phénomènes se traduisent par la
disparition des noyaux et une modification de la coloration de la fibre musculaire par
l'hémalun-éosine : coloration pâle dans les régions nécrotiques de la fibre lésée (fibres dites «
hyalines »), sombre dans les régions voisines où se situent les myofibrilles en
hypercontraction [10]. La membrane lésée laisse échapper de la fibre une partie de ses
composants et en particulier les CK.
Régénération
La régénération débute par l'activation et la multiplication des cellules satellites de la fibre
musculaire. Ces cellules prolifèrent et forment des myoblastes qui fusionnent progressivement
pour former un myotube [11]. Ces cellules sont caractérisées par des noyaux en position
centrale et une basophilie liée à leur richesse en acide ribonucléique (ARN). Au fur et à
mesure de l'enrichissement en myofibrilles, les noyaux sont repoussés en périphérie. Les
myotubes prennent progressivement les caractéristiques de myofibres matures [10]. La
persistance de noyaux centralisés est un bon marqueur de fibres régénérées.
Description habituelle
Dans les dystrophies des ceintures, la biopsie musculaire montre l'association caractéristique
nécrose/régénération. Les fibres ont un diamètre variable et présentent des internalisations
nucléaires. Des fibres lobulées peuvent être observées. Un infiltrat inflammatoire peut parfois
être présent et faire poser par erreur le diagnostic de myosite. Au début de la maladie, la
régénération compense la nécrose et le muscle garde une bonne trophicité. Le nombre de
mitoses que peut subir une cellule satellite étant limité, ce phénomène s'épuise [11]. À un
stade tardif, les fibres musculaires sont donc remplacées par de la fibrose et du tissu adipeux.
Dystrophies musculaires des ceintures autosomiques
récessives

Dans la classification actuelle, les dystrophies des ceintures autosomiques récessives sont
dénommées LGMD2. Il s'agit d'un groupe hétérogène pour lequel 15 loci sont actuellement
décrits (dont seulement 14 sont présents dans l'actuelle classification de l'European Federation
of Neurological Societies [EFNS]). Certaines formes sont fréquentes, alors que d'autres n'ont
été décrites que sur une seule famille (Tableau 2).
Calpaïnopathie (LGMD2A)
Il s'agit de la première forme de dystrophie des ceintures autosomique récessive décrite. Pour
de nombreux auteurs et en particulier pour Fardeau, à qui revient la description princeps [12],
il s'agit de la forme la plus pure de dystrophie des ceintures. La calpaïne 3 (MIM 114240)
appartient à une famille de protéases non lysosomales calcium-dépendantes. Son rôle ainsi
que ses substrats restent encore mal connus. Elle intervient dans la dégradation des
myofibrilles et des protéines du cytosquelette [13].
Épidémiologie
L'étude de la prévalence des différentes dystrophies des ceintures est difficile. L'existence
d'isolats (île de la Réunion, Pays basque, etc.) peut fortement biaiser les estimations. Selon les
études, les calpaïnopathies représentent entre 6 % et 22 % des dystrophies des ceintures [14,
15, 16, 17], dont elles sont généralement considérées comme la première étiologie.
Présentation clinique
La calpaïnopathie primaire a d'abord été décrite grâce aux études moléculaires menées sur une
communauté réunionnaise [12]. La faiblesse musculaire débute entre la première et la
troisième décennie, typiquement entre 10 et 15 ans [18, 19, 20]. Les premiers territoires
touchés se situent au niveau de la ceinture pelvienne : grand fessier et adducteurs de cuisse. Il
faut noter la préservation fréquente du quadriceps aux stades précoces. À la ceinture
scapulaire, les muscles suivants sont atteints : grand dorsal, grand pectoral, grand dentelé,
rhomboïde et biceps brachial (Figure 2A), l'atteinte du deltoïde, du triceps et des radiaux étant
tardive. Il existe fréquemment un décollement des omoplates (Figure 2B). La dissociation
entre l'amyotrophie du biceps et la conservation du triceps est typique (Figure 2A).
L'aggravation est lentement progressive mais variable, y compris à l'intérieur d'une même
famille [18]. Il peut apparaître des rétractions des tendons d'Achille généralement peu sévères.
La perte de la marche se situe entre la troisième et la quatrième décennie. Il faut noter
l'absence de pseudohypertrophie des mollets ou de macroglossie contrairement au phénotype
Duchenne/Becker présent dans de nombreuses dystrophies des ceintures. Il peut exister une
scoliose modérée et généralement non chirurgicale. La face est classiquement préservée. Il n'y
a pas d'atteinte cardiaque ni de retard intellectuel. L'insuffisance respiratoire est peu fréquente.
La durée de vie est proche de la normale [21].
Éléments paracliniques
Créatine-kinases et autres éléments biologiques
Au début de la maladie, les CK sont très élevées (jusqu'à 20 fois la normale) puis décroissent
dans un second temps. Il peut exister une hyperéosinophilie sanguine.
Imagerie musculaire

L'imagerie musculaire est très intéressante au début de la maladie en raison de la sélectivité de
l'atteinte. Il existe typiquement une atteinte précoce de la loge postérieure de cuisse avec
préservation de la loge antérieure (Figure 3) [22]. Il faut noter que les muscles restant le plus
longtemps épargnés sont : le vaste externe, le sartorius et le gracile [23]. Au niveau jambier,
on peut observer une atteinte du soléaire et du jumeau interne alors que le jumeau externe est
préservé [22]. À un stade plus évolué, l'atteinte du biceps brachial avec préservation du triceps
est évocatrice (Figure 3).
Anatomopathologie
Avec les techniques habituelles, on observe des lésions typiques de dystrophie musculaire,
avec une formule de nécrose/régénération. Bien que non spécifique, la présence de
nombreuses fibres lobulées est évocatrice. On peut parfois observer un infiltrat inflammatoire
à éosinophile pouvant faire errer le diagnostic [24]. Il est impossible d'utiliser des techniques
d'immunomarquage sur lame pour étudier la calpaïne 3. Le western blot montre une
diminution de la calpaïne 3 dans 80 % des cas [25]. Cette diminution peut toutefois être
observée dans certaines autres dystrophies telles que les dysferlinopathies et titinopathies [26,
27]. On parle alors de déficit secondaire en calpaïne 3.
Génétique moléculaire
Le locus a été identifié en 1991 grâce aux études menées sur l'île de la Réunion par Fardeau
[13, 28]. Les premières mutations du gène codant la calpaïne 3, CAPN3, ont été identifiées en
1995 [29]. D'autres isolats ont été décrits, en particulier dans le Pays basque [20]. La
recherche directe de mutations est réalisée en première intention. Toutefois, 20 % à 25 % des
patients ne présentent qu'une seule mutation [30]. Dans ce cas, une analyse de l'ARN
messager peut être réalisée. Les mutations retrouvées sont extrêmement variées en dehors des
mutations privées identifiées dans certains isolats : sud de l'île de la Réunion, Pays basque
[12, 20], etc. Sur les larges séries, environ les deux tiers des patients mutés correspondent au
phénotype décrit plus haut. Dix pour cent présentent un début précoce pouvant initialement
faire évoquer une maladie de Duchenne, 3 % un déficit de début distal et 6 % une
hypercréatine-kinasémie isolée [31].
À retenir
LGMD2A : calpaïnopathie
Origine géographique : ubiquitaire, île de la Réunion, Pays basque
Clinique
• début : 2e décennie
• prédominance : loge postérieure de cuisse
• pas d'atteinte cardiaque
• peu d'atteinte respiratoire
• perte de la marche : 4e décennie
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
1
/
29
100%