Lire l'article complet

Correspondances en Onco-Théranostic - Vol. I - n° 3 - juillet-août-septembre 2012
116
ALK
dossier thématique
Activation du récepteur ALK
dans le neuroblastome
Activation of the ALK receptor
in neuroblastoma
Isabelle Janoueix-Lerosey*, Olivier Delattre*
* Inserm U830
“Unité de génétique
et biologie des cancers”,
institut Curie, Paris.
RÉSUMÉ
Summary
»
Le neuroblastome est une tumeur pédiatrique maligne du
système nerveux sympathique périphérique ; le pronostic de ses
formes métastatiques agressives reste sombre. Des mutations
somatiques activatrices du gène ALK sont observées dans 8 %
des tumeurs sporadiques et affectent principalement 3 résidus
localisés dans le domaine kinase du récepteur. Des amplifications
géniques, aboutissant à l’activation du récepteur, ont également
été rapportées dans environ 2 % des formes sporadiques. La
démonstration du rôle clé de ces événements dans l’oncogenèse
du neuroblastome indiquait que le récepteur ALK pouvait constituer
une cible thérapeutique potentielle dans cette pathologie tumorale.
Plusieurs inhibiteurs du récepteur ALK ont été évalués dans des
modèles précliniques. Les premiers résultats d’un essai clinique de
phase I avec le crizotinib chez des enfants présentant des tumeurs
en rechute, dont des neuroblastomes, ont été présentés au congrès
de l’ASCO début juin 2012.
Mots-clés : Neuroblastome – Mutations – Prédisposition – Inhibiteurs
du récepteur ALK.
Neuroblastoma is a pediatric malignant tumor of the
peripheral sympathetic nervous system. The prognosis
of aggressive metastatic tumors remains poor. Somatic
activating mutations of the ALK gene are observed in 8%
of sporadic tumors and mainly affect 3 residues located
within the tyrosine kinase domain of the receptor. Genomic
amplifications leading to ALK activation have also been
described in about 2% of sporadic cases. The demonstration
that such events are drivers of neuroblastoma oncogenesis
has indicated that the ALK receptor may constitute a
potential target for therapy in this pediatric cancer. Several
ALK inhibitors have been evaluated in pre-clinical models.
Results of a phase I clinical trial of crizotinib for pediatric
patients with relapsed tumors including neuroblastoma
have been presented at the ASCO meeting at the beginning
of June 2012.
Keywords: Neuroblastoma – Mutations – Predisposition –
ALK inhibitors.
Mutations du gène ALK
dans le neuroblastome
En 2008, l’implication du récepteur à activité tyro-
sine kinase ALK dans le neuroblastome était révélée
à la suite de l’identification, d’une part, de mutations
somatiques activatrices du gène ALK dans des formes
sporadiques de neuroblastome et, d’autre part, de
mutations germinales dans certaines formes fami-
liales de la maladie (1-4). Les analyses fonctionnelles
démontraient par ailleurs que l’inhibition de l’activité
du récepteur dans des lignées cellulaires de neuro-
blastome s’accompagnait d’une forte réduction de la
prolifération cellulaire. En accord avec ces résultats, le
récepteur ALK était identifié indépendamment comme
une cible moléculaire dans le neuroblastome, à la suite
d’un criblage systématique de plus de 600 lignées de
cancers avec des inhibiteurs pharmacologiques de ce
récepteur (5). Ainsi était caractérisée la première cible
moléculaire dans ce cancer pédiatrique. En plus des
traitements conventionnels, la possibilité d’une thérapie
ciblée utilisant des inhibiteurs anti-ALK voyait le jour.
Mutations somatiques
Une méta-analyse portant sur une série de plus de
700 cas de neuroblastome a montré que la fréquence
des mutations somatiques du gène ALK est de l’ordre
de 8 % dans les tumeurs primaires sporadiques de

Correspondances en Onco-Théranostic - Vol. I - n° 3 - juillet-août-septembre 2012
117
Activation du récepteur ALK dans le neuroblastome
neuroblastome (6). Les mutations sont observées
dans des formes d’agressivité variable, et la grande
majorité d’entre elles affecte des acides aminés situés
dans le domaine portant l’activité tyrosine kinase du
récepteur. Deux points chauds de mutations ont été
observés en position 1174 et 1275 du récepteur ALK ; les
mutations du résidu situé en position 1245 arrivent en
troisième position (figure). Cette méta-analyse a mis en
évidence la survenue plus fréquente de la mutation en
position 1174 dans des tumeurs agressives présentant
une amplification de l’oncogène MYCN.
Mutations germinales
Trois types de mutations constitutionnelles ont été
initialement décrites dans les formes familiales de
neuroblastome, affectant les résidus G1128, R1192 et
R1275 (3, 4). La mutation la plus fréquente est observée
en position R1275. L’analyse des familles montrait que
les mutations étaient héritées et que leur pénétrance
était incomplète. Une série de patients atteints de neu-
roblastome pour lesquels une prédisposition génétique
était fortement suspectée (à savoir tumeurs diagnosti-
quées avant 1 mois de vie ou tumeurs multifocales) a
été récemment étudiée (7). Trois mutations germinales
ont été identifiées, dont 2 (T1151R and R1192P) dans
un contexte de tumeurs multifocales. Ces 2 mutations
étaient héritées de parents indemnes de la maladie.
Ces résultats confirment la diversité des mutations
germinales du gène ALK dans le neuroblastome ainsi
que leur pénétrance incomplète.
Mutations syndromiques
De façon intéressante, le spectre des mutations ger-
minales et somatiques paraissait donc différent. En
particulier, les mutations affectant les résidus F1174
et F1245 avaient été observées uniquement au niveau
somatique, ce qui suggère une potentielle létalité
embryonnaire. Plus récemment, des mutations ger-
minales de novo affectant le gène ALK, F1174V et
F1245V, ont été rapportées chez 2 patients atteints
de neuroblastome et souffrant de troubles neuro-
logiques sévères (8). Ces 2 patients présentaient, outre
un neuroblastome néonatal mutifocal, un dysfonction-
nement du tronc cérébral associé à une dysmorphie
visible à l’IRM. Ces observations impliquent le gène
ALK non seulement dans l’oncogenèse du neuro-
blastome mais également dans le développement
du système nerveux central. Elles sont en accord avec
une activation accrue du récepteur ALK induite par
certaines mutations et suggèrent l’existence d’un
effet seuil d’activation compatible avec un dévelop-
pement normal.
D’autres mécanismes pour activer
le récepteur ALK
Si les mutations somatiques du gène ALK sont obser-
vées dans moins de 10 % des neuroblastomes, plu-
sieurs autres mécanismes d’activation du récepteur
ont maintenant été identifiés. Des amplifications géno-
miques du gène ALK ont été rapportées dans environ
2 % des tumeurs primaires analysées. La majorité des
cas comportant une amplification du gène ALK pré-
sente aussi une amplification de l’oncogène MYCN. On
notera également une publication récente décrivant
une activation du récepteur ALK dans une lignée cel-
lulaire de neuroblastome présentant une amplifica-
tion du gène combinée à une délétion intragénique
aboutissant à la synthèse d’un récepteur ALK tronqué
dans son domaine extracellulaire et possédant des
propriétés oncogéniques (9). Cette situation rappelle
celle d’autres récepteurs à activité tyrosine kinase, tels
que l’EGFR et le PDGFRα, dont des délétions intragé-
niques aboutissant à la perte d’une partie du domaine
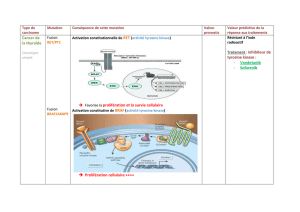
Figure. Structure du récepteur ALK, altérations somatiques observées dans le neuroblastome
et stratégies d’inhibition ciblées.
Le domaine portant l’activité tyrosine kinase est localisé dans la partie intracellulaire du récep-
teur. Les 3 mutations somatiques les plus fréquemment observées dans le neuroblastome sont
indiquées. Un premier essai clinique de phase I avec le crizotinib, un inhibiteur compétitif de
la liaison de l’ATP au récepteur ALK, a été mené aux États-Unis (17). Des expériences in vitro
indiquent que l’utilisation d’anticorps antagonistes du récepteur ALK représente potentiellement
une stratégie complémentaire d’inhibition de cette cible.
1
Domaines
MAM
Anticorps
Domaine
transmembranaire
Domaine
kinase
1030
1058
1122
F1174
F1245
R1275
1620
1376
Inhibiteurs
– crizotinib : phase I
– autres : préclinique
Amplifications
2-4 % des tumeurs
Mutations
8 % des tumeurs

Correspondances en Onco-Théranostic - Vol. I - n° 3 - juillet-août-septembre 2012
118
dossier thématique
ALK
extracellulaire sont observées dans plusieurs types
de cancer et confèrent un pouvoir transformant aux
récepteurs variants. Dans le cas du récepteur ALK, la
récurrence de ce type de délétion du domaine extra-
cellulaire semble néanmoins très faible. De façon plus
significative, l’étude de Schulte et al. montre que le
profil transcriptomique des tumeurs présentant une
forte expression du récepteur ALK ressemble à celui
des tumeurs avec amplification ou mutation du gène
ALK et que les patients correspondants présentent
un pronostic défavorable (10). La méta-analyse de De
Brouwer et al. a également mis en évidence un mauvais
pronostic des patients dont les tumeurs présentent un
niveau élevé d’expression du transcrit ALK (6).
Inhibition du récepteur ALK
Données précliniques
Plusieurs études ont démontré que l’inhibition du
récepteur ALK par ARN interférence ou molécules
pharmacologiques induit une forte diminution de la
prolifération cellulaire dans des lignées cellulaires de
neuroblastome ou des modèles hétérologues dans
lesquels le récepteur ALK est surexprimé. Dans des
lignées de neuroblastome, une telle inhibition a été
rapportée non seulement dans les cas avec ampli-
fication ou mutation du gène ALK mais également
dans certaines lignées présentant un récepteur ALK
non altéré. Le travail de Passoni et al. suggère que le
récepteur ALK sauvage exprimé au-dessus d’un seuil
critique aboutissant à son activation oncogénique
pourrait être ciblé par des inhibiteurs spécifiques (11).
Par conséquent, le récepteur ALK constitue une cible
thérapeutique dans le neuroblastome, dans un pour-
centage de cas probablement supérieur à celui des
seuls cas avec mutations activatrices et/ou amplifica-
tion. L’identification de biomarqueurs spécifiques et
fiables permettant de repérer l’ensemble des patients
qui pourraient bénéficier d’une thérapie ciblée anti-
ALK reste néanmoins un challenge.
Plusieurs inhibiteurs pharmacologiques du récepteur
ALK ont montré une activité dans des modèles précli-
niques de neuroblastome. Nous citerons entre autres le
TAE684, spécifique du récepteur ALK, le GSK1838705A,
ciblant les récepteurs ALK et IGF-1R, et le crizotinib,
ciblant les oncogènes ALK et c-Met (12). Néanmoins,
il est apparu rapidement que la sensibilité à ces inhi-
biteurs était variable selon la mutation modifiant le
récepteur. En particulier, la forme ALK mutée F1174L
est moins sensible au crizotinib que la forme mutée
R1275Q (13). Ces données suggèrent donc que le
développement de plusieurs inhibiteurs anti-ALK sera
nécessaire pour traiter de façon efficace les patients
atteints de neuroblastome avec mutations du gène
ALK. Cela est d’autant plus vrai que des résistances au
crizotinib ont d’ores et déjà été rapportées chez des
adultes présentant des cancers avec des protéines de
fusion ALK et traités avec cette molécule. Ainsi, 1 cas
de tumeur myofibroblastique inflammatoire présentant
une mutation F1174L du gène ALK après traitement
avec l’inhibiteur anti-ALK a récemment été décrit (14).
L’utilisation d’anticorps dirigés contre le récepteur ALK
représente une approche alternative et potentiellement
complémentaire d’inhibition de ce récepteur. En effet,
il a été récemment montré, in vitro, qu’un anticorps
monoclonal antagoniste induisait une inhibition de
la prolifération et une cytotoxicité cellulaire de lignées
de neuroblastome présentant ou non une mutation du
récepteur ALK (15, 16).
Efficacité du crizotinib chez des patients
atteints de neuroblastome
De façon remarquable, un essai clinique de phase I a
commencé aux États-Unis avec le crizotinib en 2009,
soit 18 mois après la publication des travaux identifiant
le récepteur ALK comme une cible thérapeutique dans
le neuroblastome, chez des enfants atteints de tumeurs
solides ou de lymphomes anaplasiques à grandes
cellules en rechute. Lors du dernier congrès annuel de
l’American Society of Clinical Oncology (ASCO®), qui s’est
tenu début juin 2012 à Chicago, le Dr Y.P. Mosse a rap-
porté les résultats suivants : la toxicité du crizotinib s’est
avérée minime chez les patients traités ; 2 rémissions
complètes et 1 maladie stable prolongée ont été obser-
vées à la suite d’un traitement par le crizotinib dans un
groupe de 8 patients atteints de “NB-ALK positif”, dont
2 présentant une mutation germinale ; une rémission
complète et 6 maladies stables prolongées ont été
observées dans un groupe de 19 patients atteints de “NB
non-ALK positif” (17). Ces premiers résultats indiquent
que certains patients atteints de neuroblastome en
rechute pourraient bénéficier d’un traitement par cri-
zotinib. À noter que, dans le cas des lymphomes ana-
plasiques, la réponse au crizotinib semblait meilleure,
avec 7 patients en rémission complète sur 8 traités.
Conclusion
Des progrès majeurs ont été obtenus ces dernières
années dans la compréhension de la biologie du
neuro blastome. Ainsi, la mise en évidence de l’acti-
vation du récepteur ALK chez certains patients atteints

Correspondances en Onco-Théranostic - Vol. I - n° 3 - juillet-août-septembre 2012
119
Activation du récepteur ALK dans le neuroblastome
de neuroblastome a ouvert la voie d’une médecine
personnalisée utilisant des inhibiteurs spécifiques
de ce récepteur. Des résultats encourageants ont été
obtenus récemment avec le crizotinib. Néanmoins,
certaines questions cruciales restent à élucider : quelles
méthodes doit-on utiliser pour mettre en évidence
l’activation du récepteur ALK ? Quels patients pour-
raient bénéficier d’une thérapie anti-ALK ? Les efforts
de recherche doivent donc être poursuivis afin de
comprendre sur le plan fondamental la biologie du
récepteur ALK et de définir ensuite les meilleures
options thérapeutiques. ■
1.
Chen Y, Takita J, Choi YL et al. Oncogenic mutations of ALK
kinase in neuroblastoma. Nature 2008;455:971-4.
2. George RE, Sanda T, Hanna M et al. Activating mutations
in ALK provide a therapeutic target in neuroblastoma. Nature
2008;455:975-8.
3. Janoueix-Lerosey I, Lequin D, Brugieres L et al. Somatic and
germline activating mutations of the ALK kinase receptor in
neuroblastoma. Nature 2008;455:967-70.
4.
Mosse YP, Laudenslager M, Longo L et al. Identification of
ALK as a major familial neuroblastoma predisposition gene.
Nature 2008;455:930-5.
5. McDermott U, Iafrate AJ, Gray NS et al. Genomic altera-
tions of anaplastic lymphoma kinase may sensitize tumors
to anaplastic lymphoma kinase inhibitors. Cancer Res
2008;68:3389-95.
6. De Brouwer S, De Preter K, Kumps C et al. Meta-analysis
of neuroblastomas reveals a skewed ALK mutation spec-
trum in tumors with MYCN amplification. Clin Cancer Res
2010;16:4353-62.
7.
Bourdeaut F, Ferrand S, Brugieres L et al. ALK germline muta-
tions in patients with neuroblastoma: a rare and weakly pene-
trant syndrome. Eur J Hum Genet 2012;20:291-7.
8. De Pontual L, Kettaneh D, Gordon CT et al. Germline gain-
of-function mutations of ALK disrupt central nervous system
development. Hum Mutat 2011;32:272-6.
9. Okubo J, Takita J, Chen Y et al. Aberrant activation of ALK kinase
by a novel truncated form ALK protein in neuroblastoma. Oncogene
2012 Jan 16. doi: 10.1038/onc.2011.616 [Epub ahead of print].
10. Schulte JH, Bachmann HS, Brockmeyer B et al. High ALK
receptor tyrosine kinase expression supersedes ALK mutation as
a determining factor of an unfavorable phenotype in primary
neuroblastoma. Clin Cancer Res 2011;17:5082-92.
11. Passoni L, Longo L, Collini P et al. Mutation-independent
anaplastic lymphoma kinase overexpression in poor prognosis
neuroblastoma patients. Cancer Res 2009;69:7338-46.
12. Zou HY, Li Q, Lee JH et al. An orally available small-molecule
inhibitor of c-Met, PF-2341066, exhibits cytoreductive anti-
tumor efficacy through antiproliferative and antiangiogenic
mechanisms. Cancer Res 2007;67:4408-17.
13. Bresler SC, Wood AC, Haglund EA et al. Differential inhibitor
sensitivity of anaplastic lymphoma kinase variants found in
neuroblastoma. Sci Transl Med 2011;3:108ra14.
14. Sasaki T, Okuda K, Zheng W et al. The neuroblastoma
associated F1174L ALK mutation causes resistance to an
ALK kinase inhibitor in ALK translocated cancers. Cancer Res
2010;70:10038-43.
15.
Carpenter EL, Haglund EA, Mace EM et al. Antibody tar-
geting of anaplastic lymphoma kinase induces cytotoxicity of
human neuroblastoma. Oncogene 2012 Jan 23. doi: 10.1038/
onc.2011.647 [Epub ahead of print].
16. Moog-Lutz C, Degoutin J, Gouzi JY et al. Activation and
inhibition of anaplastic lymphoma kinase receptor tyrosine
kinase by monoclonal antibodies and absence of agonist
activity of pleiotrophin. J Biol Chem 2005;280:26039-48.
17. Mosse YP, Balis FM, Lim MS et al. Efficacy of crizotinib in
children with relapsed/refractory ALK-driven tumors inclu-
ding anaplastic large cell lymphoma and neuroblastoma: A
Children’s Oncology Group phase I consortium study. J Clin
Oncol 2012;30(suppl.):abstr 9500.
Références
Périodique de formation
www.edimark.fr
Société éditrice : EDIMARK SAS
CPPAP : 0514 T 91341 – ISSN : 2259-6674
Trimestriel
Prix du numéro : 36 €
Vol. I – n° 2
Avril-mai-juin 2012
A
bonnez-vous, page 66
DOSSIER
Marqueurs prédictifs
de sensibilité
et de résistance
aux anti-HER
Coordonnatrice : K. Leroy
Éditorial
De l’orage
dans l’ HER…
Fiche technique
Score HER2
pour les carcinomes gastriques
Échos des congrès
AACR 2012
SPÉCIAL
ABONNEMENT
CONGRÈS
Voir page 120
-50
%
Éditorial
ALK en oncologie :
un gène, plusieurs altérations,
des maladies différentes,
une cible
Cas clinique
Tests moléculaires
et CCR métastatique
chez un sujet jeune
Échos des congrès
- Highlights de l’ASCO
- Pathologie moléculaire
des tumeurs : 1
re
réunion
de restitution des résultats
Périodique de formation
www.edimark.fr
Société éditrice : EDIMARK SAS
CPPAP : 0514 T 91341 – ISSN : 2259-6674
Trimestriel
Prix du numéro : 36 €
Vol. I – n° 3
Juillet-août-sept. 2012
A
bonnez-vous, page 120
DOSSIER
ALK
Coordonnatrice :
A. Vincent-Salomon
Au sommaire du prochain numéro
Dossier thématique :
Réparation
Coordonné par Frédérique Penault-Llorca,
avec la participation de : Julian Biaud,
Sabrina Boyrie, Emmanuel Chautard,
Jean-François Emile, Jonathan Khalifa,
Ken Olaussen, Nicolas Sevenet,
Tony Sourisseau...
» Parution en décembre 2012
www.edimark.fr
Abonnez-vous
sur
1
/
4
100%