BRAF et potentiel thérapeutique dans les tumeurs humaines -activating mutations and therapeutic developments

Correspondances en Onco-Théranostic - Vol. I - n° 1 - janvier-février-mars 2012
30
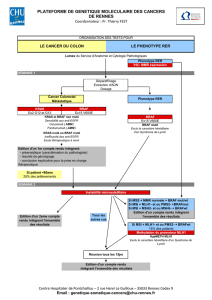
BRAF en oncologie
dossier thématique
Mutations activatrices de BRAF
et potentiel thérapeutique
dans les tumeurs humaines
BRAF-activating mutations and therapeutic developments
in human malignancies
Olivier Mir*
RÉSUMÉ
Summary
»
Les mutations activatrices de BRAF pourraient permettre de
définir un groupe de tumeurs ayant une sensibilité particulière aux
récents inhibiteurs de BRAF. Le rôle de ces mutations pour prédire
la sensibilité au vémurafénib dans les mélanomes métastatiques
semble établi, mais reste incertain pour d’autres sous-types
tumoraux.
»
Les données récentes du développement clinique d’autres
inhibiteurs de BRAF semblent indiquer que cette nouvelle classe
de médicaments anticancéreux pourrait avoir un large spectre
d’activité. Il reste cependant à établir si cette activité clinique est liée
à l’inhibition de BRAF en elle-même ou à l’inhibition concomitante
d’autres cibles.
Mots-clés : MAP-kinases – BRAF – Cancer – Mutation – Mélanome
– Antiangiogéniques.
BRAF-activating mutations may contribute to identify a
group of tumours with a particular sensitivity to recent BRAF
inhibitors. The role of such mutations in the prediction of
response to vemurafenib in metastatic melanoma seems
established, but remains to be demonstrated for other tumour
types.
Recent data from early clinical trials of BRAF inhibitors suggest
that this new class of anti-cancer drugs could have a broad
spectrum of anti-tumour activity. Whether clinical activity
is underlied by BRAF inhibition per se, or by the inhibition of
other targets remains to be determined.
Keywords: MAP-kinases – BRAF – Cancer – Mutation –
Melanoma – Angiogenesis inhibitors.
L’
intérêt pour le développement clinique d’inhi-
biteurs de BRAF n’est pas nouveau, même si
les données disponibles dans le traitement
des mélanomes sont récentes (cf. article de C. Robert
et C. Mateus, p. 24).
Historiquement, le développement thérapeutique
d’inhibiteurs de la voie de signalisation de RAF a suivi
la découverte du fait que RAF constituait un effecteur
direct de RAS et de récepteurs de facteurs de croissance
situés en amont de la voie des MAP-kinases, tels que
le récepteur à l’EGF (1, 2). Par ailleurs, la découverte de
mutations activatrices de BRAF (dans environ 7 % des
tumeurs solides, tous types confondus ; cf. article de
J.F. Émile, p. 20) a renforcé l’intérêt pour ces inhibiteurs
dans les mélanomes métastatiques (près de 50 % des
tumeurs), mais aussi dans les carcinomes papillaires de
la thyroïde (40 à 50 %) et les cancers colorectaux (10 %)
[3, 4].
Le sorafénib : premier inhibiteur
historique de BRAF
Le premier inhibiteur de BRAF dont l’activité cli-
nique a été bien établie est le sorafénib (BAY43-9006,
Nexavar®, Bayer Healthcare), développé à la fin des
années 1990. Quatre essais de phase I ont permis de
fixer la dose recommandée pour les essais de phase II
(RP2D) à 400 mg × 2/j par voie orale, l’augmentation
de l’exposition plasmatique au-delà de la RP2D étant
infraproportionnelle à la dose (5). Les principaux effets
indésirables observés étaient la diarrhée et la fatigue de
grade 3 à 800 mg × 2/j, et la toxicité cutanée de grade
3 à 600 mg 2 × 2/j (5).
Le fait que le sorafénib inhibe non seulement BRAF et
CRAF, mais aussi les récepteurs au PDFG et au VEGF, a
orienté son développement vers des indications où la
voie du VEGF semblait critique, en particulier le car-
* Oncologie médicale et
pharmacologie clinique,
groupe hospitalier Cochin-
Broca-Hôtel-Dieu, AP-HP ;
université Paris-Descartes,
Sorbonne Paris Cité.

Correspondances en Onco-Théranostic - Vol. I - n° 1 - janvier-février-mars 2012
31
Mutations activatrices de BRAF et potentiel thérapeutique dans les tumeurs humaines
cinome hépatocellulaire (CHC) et les cancers du rein
métastatiques. Le sorafénib est à ce jour le seul traite-
ment pharmacologique approuvé pour le traitement
des CHC avancés ou métastatiques (6). Il est par ailleurs
indiqué dans le traitement des cancers du rein avancés
après échec d’un traitement préalable par cytokines (7),
bien que des inhibiteurs plus spécifiques de la voie du
VEGF semblent plus efficaces en deuxième ligne dans
cette indication (8).
Les données d’activité clinique obtenues en phase II
sont également très prometteuses dans les cancers de
la thyroïde différenciés (9, 10) et les ostéosarcomes (11),
mais plus mitigées dans d’autres types tumoraux tels
que les cancers bronchiques non à petites cellules et les
sarcomes des tissus mous (12, 13) [tableau I]. Le fait que
les caractéristiques moléculaires des tumeurs n’étaient
pas prises en compte lors du choix des patients pourrait
en partie expliquer ces résultats décevants. Par ailleurs,
le sorafénib est caractérisé par une grande variabilité
pharmacocinétique interindividuelle, ce qui suggère
que certains patients pourraient être sous-exposés à
la dose recommandée de 400 mg × 2/j (14). Par ailleurs,
des résultats récents suggèrent également une variabi-
lité intra-individuelle, avec une baisse de l’exposition au
fil du temps (15), et une escalade de dose pourrait être
nécessaire au moment de la progression (16).
Enfin, l’activité du sorafénib dans ces différentes patho-
logies pourrait reposer sur l’inhibition d’autres cibles
que BRAF. À titre d’illustration, on citera l’échec de son
développement dans les mélanomes métastatiques
et l’absence de corrélation entre l’activité et le statut
mutationnel de BRAF dans cette indication (17, 18).
Le vémurafénib :
mélanomes métastatiques, et au-delà ?
Le vémurafénib (PLX4032, R05185426, Zelboraf®, Roche)
est un inhibiteur plus spécifique de BRAF, dont le déve-
loppement en phase I a permis de définir une RP2D à
TableauI. Données d’activité (essais de phase II) du sorafénib dans diverses tumeurs solides.
Type tumoral n Taux de réponse (%) Stabilisations (%) Survie sans progression (mois) Survie globale (mois)
Cancers différenciés de la thyroïde (9) 41 15,0 56,0 15,0 (IC95 : 10,0-27,5) 23 (IC95 : 18,0-34,0)
Cancers médullaires de la thyroïde sporadiques (10) 16 6,3 87,5 17,9 (IC95 : 8,0-non atteint) Non atteinte
Sarcomes des tissus mous (13) 122 4,9 50,8 3,2 (IC95 : 2,5-3,7) 14,3 (IC95 : 12,2-19,2)
Ostéosarcomes (11) 35 14,0 34,0 4,0 (IC95 : 2,0-5,0) 7 (IC95 : 7,0-8,0)
Cancers bronchiques non à petites cellules (12) 25 12,0 24,0 2,8 (IC95 : 1,8-4,6) 8,8 (IC95 : 5,1-16,9)
TableauII. Vémurafénib : essais cliniques en cours (21).
Identifiant Titre
de l’étude Type
d’étude Tumeurs
concernées Critère principal
dejugement Critères secondaires
dejugement
NCT01495988 Trial of vemurafenib with
or without bevacizumab
in patients with stage IV
BRAFV600 mutant mela-
noma
Phase II randomisée,
vémurafénib 960 mg × 2/j
vs vémurafénib 960mg
× 2/j + bévacizumab
15mg/kg/3 semaines
Mélanomes BRAFV600
mutés
Survie sans
progression
Effets indésirables,
survie globale,
biomarqueurs
d’efficacité
NCT01531361 Vemurafenib and sorafe-
nib in advanced cancer
Phase I, vémurafénib
(dose initiale : 240 mg
× 2/j) + sorafénib (dose
initiale : 200 mg × 2/j)
Tumeurs BRAFV600
mutées
Dose maximale
tolérée
Taux de réponse
à8semaines
NCT01524978 A study of vemurafenib in
patients with BRAF V600
mutation-positive cancers
Phase II, vémurafénib
960 mg × 2/j
Tumeurs solides
et myélomes BRAFV600
mutés, sauf mélanomes
et cancers papillaires
de la thyroïde
Taux de réponse
à8semaines
Survie sans progression,
survie globale, taux
deréponse à 3 mois,
tempsjusqu’à progres-
sion, durée de réponse,
effetsindésirables
NCT01286753 A study of RO5185426 in
patients with metastatic
or unresectable papillary
thyroid cancer positive for
the BRAF V600 mutation
Phase II, vémurafénib
960 mg × 2/j
Cancers papillaires
de la thyroïde BRAFV600
mutés
Taux de réponse Survie sans progression,
survie globale,
durée deréponse,
effets indésirables selon
prétraitement par sorafénib ;
pharmacocinétique

Correspondances en Onco-Théranostic - Vol. I - n° 1 - janvier-février-mars 2012
32
BRAF en oncologie
dossier thématique
960 mg × 2/j (par voie orale), avec un profil de toxicité
semblant plus favorable que celui du sorafénib (19). Son
développement récent dans le traitement des méla-
nomes métastatiques BRAFV600 mutés est détaillé dans
l’article de C. Robert et C. Mateus, p. 24. On notera que le
vémurafénib est caractérisé par une importante variabi-
lité pharmacocinétique interindividuelle (19), l’aire sous
la courbe concentration-temps de 0 à 24 heures étant
en moyenne de 1 741 ± 639 μmol/l.h, ce qui suggère,
là encore, que certains patients pourraient être sous-
exposés à la dose standard de 960 mg × 2/j.
Son activité est en cours d’évaluation, en combinai-
son avec d’autres agents (notamment les anti-VEGF)
et en monothérapie dans les tumeurs solides (dont
les cancers papillaires de la thyroïde, qui font l’objet
d’une étude dédiée) BRAFV600 mutés (tableau II, p. 31).
De façon notable, les inhibiteurs de RAF peuvent exer-
cer un effet paradoxal (activation de la voie des MAP-
kinases) dans les tumeurs BRAF sauvages mais porteuses
de mutations de RAS (20), ce qui souligne l’importance
de la caractérisation des tumeurs au niveau molécu-
laire avant traitement. Cet effet semble aussi expliquer
l’apparition de tumeurs cutanées (kératoacanthomes/
carcinomes épidermoïdes) sous traitement (21).
Autres inhibiteurs de BRAF :
moins sélectifs, aussi prometteurs
Si les bons résultats du vémurafénib dans les méla-
nomes métastatiques porteurs de la mutation V600 de
BRAF sont indéniables, les premiers résultats d’autres
inhibiteurs moins sélectifs de BRAF sont également
prometteurs.
✓
Le RAF265 (Novartis) est un inhibiteur multikinases
ciblant BRAF mais aussi CRAF, C-Kit, VEGFR2 et PDGFRβ.
Trente-neuf patients atteints de mélanomes BRAFV600
mutés, mais aussi 27 patients atteints de mélanomes
BRAFV600 sauvages, ont été traités dans un essai de
phase I dont les résultats ont été présentés au congrès
de l’ASCO en 2011 (22). Les principales toxicités limitant
la dose étaient l’élévation de la lipase, la diarrhée, la
toxicité rétinienne et un épisode d’embolie pulmonaire.
Sept cas de thrombopénie de grade 3-4 ont également
été rapportés. La dose maximale tolérée était de 48 mg/j
par voie orale. Les auteurs ont cependant rapporté
6 réponses partielles et 1 réponse complète. De façon
intéressante, 2 des réponses partielles et la réponse
complète étaient observées chez des patients dont la
tumeur était BRAFV600 sauvage. Une cohorte d’exten-
sion de cette étude (NCT00304525) est actuellement
ouverte (23).
✓
Le RO5126766 (Roche) est un inhibiteur mixte de RAF
et de MEK qui a été étudié dans un essai de phase I chez
52 patients (21 mélanomes, 10 cancers colorectaux)
[24]. La dose maximale tolérée était de 2,7 mg/j per
os, 1 semaine sur 2. Les principaux effets indésirables
incluaient le rash, le flou visuel, la diarrhée et la toxi-
cité musculaire (myosites cliniques et biologiques).
L’activité antitumorale de cet inhibiteur mixte de RAF
et de MEK était encourageante, avec 3 réponses chez
des patients atteints de mélanomes (dont 2 BRAFV600
mutés et 1 NRAS muté) et 9 stabilisations (mélanomes
et cancers colorectaux).
✓
Par analogie, le GSK2118436 (inhibiteur de RAF,
GlaxoSmithKline) a été associé à un inhibiteur de MEK
(GSK1120212) dans un essai de phase I chez 109 patients
porteurs de tumeurs BRAFV600 mutées (dont 101 cas
de mélanome) [25]. Le profil de toxicité comportait
rash, diarrhée et asthénie. Les auteurs ont rapporté
47 réponses chez 71 patients naïfs de traitement anti-
BRAF, ainsi que 3 réponses partielles et 12 stabilisations
chez les 24 patients prétraités par un inhibiteur de BRAF.
Ces résultats suggèrent que l’inhibition combinée de
BRAF et de MEK permettrait de contourner certains
mécanismes de la résistance aux inhibiteurs de BRAF.
Le GSK2118436 est actuellement en cours de déve-
loppement en phase II, notamment dans les cancers
bronchiques non à petites cellules (NCT01336634), les
mélanomes (NCT01153763) et d’autres types tumoraux
(NCT01231594), tous BRAFV600 mutés.
✓Le régorafénib (BAY 73-4506, Bayer Healthcare) est
un autre inhibiteur multikinases ciblant BRAF (sauvage
et muté), CRAF, C-Kit, RET, TIE-2, VEGFR2-3, FGFR1,
PDGFRβ et MAP-kinase p38 (26).
Deux schémas d’administration ont été déterminés
en phase I : 160 mg/j 3 semaines sur 4 et 100 mg/j en
continu (27, 28). De façon remarquable, 2 réponses
partielles RECIST ont été observées avec le premier
schéma (1 cancer du rein et 1 ostéosarcome). Les toxi-
cités limitant la dose étaient la fièvre, l’hyperkératose
palmoplantaire (assez semblable à celle observée sous
sorafénib) et la leucopénie. Les effets indésirables
incluaient également la diarrhée, le rash, l’asthénie et
l’hypertension artérielle.
Les données de phase II sont prometteuses, en par-
ticulier dans les cancers du rein (non traités au pré-
alable) [29], avec 31 % de réponses partielles et 50 %
de stabilisations. Des résultats intéressants (69 % de
stabilisations) ont également été observés en deuxième
ligne chez des patients atteints de CHC prétraités par
sorafénib (30). Des études de phase II et III sont en
cours dans divers types tumoraux (cancers colorec-
taux notamment), en monothérapie ou en association

Correspondances en Onco-Théranostic - Vol. I - n° 1 - janvier-février-mars 2012
33
Mutations activatrices de BRAF et potentiel thérapeutique dans les tumeurs humaines
avec des schémas de chimiothérapie conventionnels
(NCT01103323, NCT01298570, NCT01289821).
Enfin, une étude de phase II récente a évalué le
schéma intermittent (160 mg/j, 3 semaines sur 4) chez
34 patients atteints de GIST en progression après un
traitement par imatinib et sunitinib (31). Dix-neuf des
22 patients évaluables avaient une maladie contrô-
lée (réponse ou stabilisation) après 16 semaines de
traitement. Les réponses et les stabilisations étaient
observées chez des patients dont la tumeur portait ou
non des mutations des exons 9 ou 11 de KIT. Un essai de
phase III randomisé contre placebo (NCT01271712) est
en cours de finalisation, et pourrait confirmer la place du
régorafénib en troisième ligne de traitement des GIST.
Des résultats préliminaires indiquent par ailleurs que les
inhibiteurs de BRAF en monothérapie ont une activité
limitée dans les cancers coliques BRAF mutés, probable-
ment du fait de l’activation d’EGFR, suggérant que la
combinaison d’un inhibiteur de BRAF et d’un inhibiteur
d’EGFR permettrait de lever cette résistance (32).
Conclusion
Les inhibiteurs de BRAF, plus ou moins sélectifs, repré-
sentent une classe émergente de thérapeutiques anti-
cancéreuses non seulement dans le traitement des
mélanomes BRAF
V600
mutés, mais aussi dans de nom-
breuses autres indications. Démontrer que l’activité
du médicament est liée à l’inhibition de BRAF et non à
celle d’une ou de plusieurs autres cibles est cependant
délicat, et l’optimisation du développement ultérieur
de ces drogues passera nécessairement par des études
de biomarqueurs tumoraux, mais aussi par des études
pharmacocliniques, compte tenu du profil pharmaco-
cinétique de ces médicaments.
■
Conitd’intérêts. Le Dr O. Mir est membre d’un advisory board
pour les laboratoires Roche et a été consultant ponctuel pour les
laboratoires Roche, Bayer, Pfizer et Servier.
L’auteur n’a pas de conflit d’intérêts en rapport direct avec le pré-
sent travail.
1.
Chong H, Vikis HG, Guan KL. Mechanisms of regulating the
Raf kinase family. Cell Signal 2003;15:463-9.
2.Mercer KE, Pritchard CA. Raf proteins and cancer: B-Raf
is identified as a mutational target. Biochim Biophys Acta
2003;1653:25-40.
3.Davies H, Bignell GR, Cox C et al. Mutations of the BRAF
gene in human cancer. Nature 2002;417:949-54.
4.Kimura ET, Nikiforova MN, Zhu Z et al. High prevalence of
BRAF mutations in thyroid cancer: genetic evidence for consti-
tutive activation of the RET/PTC-RAS-BRAF signaling pathway
in papillary thyroid carcinoma. Cancer Res 2003;63:1454-7.
5.Strumberg D, Clark JW, Awada A et al. Safety, pharmacoki-
netics, and preliminary antitumor activity of sorafenib: a review
of four phase I trials in patients with advanced refractory solid
tumors. Oncologist 2007;12:426-37.
6.Llovet JM, Ricci S, Mazzaferro V et al. Sorafenib in advanced
hepatocellular carcinoma. N Engl J Med 2008;359:378-90.
7.Escudier B, Eisen T, Stadler WM et al. Sorafenib in advanced
clear-cell renal-cell carcinoma. N Engl J Med 2007;356:125-34.
8.Rini BI, Escudier B, Tomczak P et al. Comparative effectiveness
of axitinib versus sorafenib in advanced renal cell carcinoma
(AXIS): a randomised phase 3 trial. Lancet 2011;378:1931-9.
9.Kloos RT, Ringel MD, Knopp MV et al. Phase II trial of sorafenib
in metastatic thyroid cancer. J Clin Oncol 2009;27:1675-84.
10.Lam ET, Ringel MD, Kloos RT et al. Phase II clinical trial
of sorafenib in metastatic medullary thyroid cancer. J Clin
Oncol 2010;28:2323-30.
11.Grignani G, Palmerini E, Dileo P et al. A phase II trial of
sorafenib in relapsed and unresectable high-grade osteosar-
coma after failure of standard multimodal therapy: an Italian
Sarcoma Group study. Ann Oncol 2011;23:508-16.
12.Dy GK, Hillman SL, Rowland KM Jr. et al. A front-line win-
dow of opportunity phase 2 study of sorafenib in patients with
advanced nonsmall cell lung cancer: North Central Cancer
Treatment Group Study N0326. Cancer 2011;116:5686-93.
13.
Maki RG, D’Adamo DR, Keohan ML et al. Phase II study of
sorafenib in patients with metastatic or recurrent sarcomas.
J Clin Oncol 2009;27:3133-40.
14.Hornecker M, Blanchet B, Billemont B et al. Saturable
absorption of sorafenib in patients with solid tumors: a popu-
lation model. Invest New Drugs 2011 Oct 18 [Epub ahead
of print].
15.Arrondeau J, Mir O, Boudou-Rouquette P et al. Sorafenib
exposure decreases over time in patients with hepatocellular
carcinoma. Invest New Drugs 2011 Oct 29 [Epub ahead of
print].
16.
Semrad TJ, Gandara DR, Lara PN, Jr. Enhancing the clinical
activity of sorafenib through dose escalation: rationale and
current experience. Ther Adv Med Oncol 2011;3:95-100.
17.Egberts F, Kahler KC, Livingstone E, Hauschild A. Metastatic
melanoma: scientific rationale for sorafenib treatment and
clinical results. Onkologie 2008;31:398-403.
18.Ott PA, Hamilton A, Min C et al. A phase II trial of sorafe-
nib in metastatic melanoma with tissue correlates. PLoS One
2011;5:e15588.
19.Flaherty KT, Puzanov I, Kim KB et al. Inhibition of muta-
ted, activated BRAF in metastatic melanoma. N Engl J Med
2010;363:809-19.
20.
Heidorn SJ, Milagre C, Whittaker S et al. Kinase-dead BRAF
and oncogenic RAS cooperate to drive tumor progression
through CRAF. Cell 2010;140:209-21.
21.Cox AD, Der CJ. The RAF inhibitor paradox revisited. Cancer
Cell 2012;21:147-9.
22.Sharfman WH, Hodi FS, Lawrence DP et al. Results from
the first-in-human (FIH) phase I study of the oral RAF inhibitor
RAF265 administered daily to patients with advanced cuta-
neous melanoma. Proc Am Soc Clin Oncol 2011. J Clin Oncol
2011;29(Suppl.):abstract 8508.
23.http://clinicaltrials.gov
24.Dolly SO, Albanell J, Kraeber-Bodere F et al. First-in-human,
safety, pharmacodynamic (PD) and pharmacokinetic (PK)
trial of a first-in-class dual RAF/MEK inhibitor, RO5126766, in
patients with advanced or metastatic solid tumors. Proc Am
Soc Clin Oncol 2011. J Clin Oncol 2011;29(Suppl.):abstract 3006.
25.
Infante JR, Falchook GS, Lawrence DP et al. Phase I/II study
to assess safety, pharmacokinetics, and efficacy of the oral MEK
1/2 inhibitor GSK1120212 (GSK212) dosed in combination with
the oral BRAF inhibitor GSK2118436 (GSK436). Proc Am Soc Clin
Oncol 2011. J Clin Oncol 2011;29(Suppl.):abstract CRA8503.
26.Wilhelm SM, Dumas J, Adnane L et al. Regorafenib (BAY
73-4506): a new oral multikinase inhibitor of angiogenic,
stromal and oncogenic receptor tyrosine kinases with potent
preclinical antitumor activity. Int J Cancer 2011;129:245-55.
27.
Frost A, Buechert M, Unger C et al. Phase I study of BAY
73-4506, an inhibitor of oncogenic and angiogenic kinases,
in patients with advanced solid tumors: final results of a dose-
escalation study. Proc Am Soc Clin Oncol 2008. J Clin Oncol
2008;26(Suppl.):abstract 2558.
28.
Shimizu T, Tolcher AW, Patnaik A et al. Phase I dose-esca-
lation study of continuously administered regorafenib (BAY
73-4506), an inhibitor of oncogenic and angiogenic kinases, in
patients with advanced solid tumors. Proc Am Soc Clin Oncol
2010. J Clin Oncol 2010;28(Suppl.):abstract 3035.
29.
Eisen T, Joensuu H, Nathan P et al. Phase II trial of the oral
multikinase inhibitor regorafenib (BAY 73-4506) as first-line
therapy in patients with metastatic or unresectable renal cell
carcinoma (RCC). Eur J Cancer 2011;47(Suppl.):abstract 7141.
30.Bolondi L, Tak WY, Gasbarrini A et al. Phase II safety study
of the oral multikinase inhibitor regorafenib (BAY 73-4506) as
second-line therapy in patients with hepatocellular carcinoma.
Eur J Cancer 2011;7:abstract 6576.
31.George S, Von Mehren M, Heinrich MC et al. A multicenter
phase II study of regorafenib in patients (pts) with advanced
gastrointestinal stromal tumor (GIST), after therapy with ima-
tinib (IM) and sunitinib (SU). Proc Am Soc Clin Oncol 2011.
J Clin Oncol 2011;29(Suppl.):abstract 10007.
32.Prahallad A, Sun C, Huang S et al. Unresponsiveness of
colon cancer to BRAF(V600E) inhibition through feedback
activation of EGFR. Nature 2012;483:100-3.
Références
1
/
4
100%