Epissage alternatif dans le génome humain: variants fonctionnels ou

Proposition sujet recherche M1 ou M2 2015-2016!
!
Epissage(alternatif(dans(le(génome(humain:(variants(fonctionnels(ou(bien(erreurs(
de(la(machinerie(d'épissage?
Directeur: Laurent DURET (Laurent.Duret@univ-lyon1.fr, LBBE – UMR 5558)
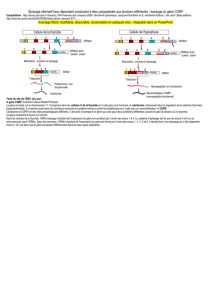
Chez les mammifères, la très grande majorité des gènes sont soumis à des phénomènes d'épissage
alternatif (rétention d'introns, saut d'exons, …). De nombreuses observations indiquent que ce
processus peut contribuer à accroitre le répertoire fonctionnel des génomes, et permet également de
réguler le niveau d'expression de certains gènes. Cependant, d'autres indices suggèrent qu'une
fraction significative des variants d'épissage détectés dans un transcriptome pourrait en fait
correspondre à des transcrits non-fonctionnels, résultant simplement d'erreurs de la machinerie
d'épissage.
Parmi tous les variants d'épissage qui sont produits par un organisme, quelle fraction correspond à
des erreurs (couteuses pour l'organisme) et quelle fraction correspond à des variants fonctionnels
(utiles à l'organisme)? Cette question reste aujourd'hui encore largement débattue dans la
communauté scientifique.
Pour y répondre, nous proposons d'analyser les signaux qui régulent l'efficacité de l'épissage des
introns (en particulier les sites donneurs et accepteurs d'épissage) pour déterminer les contraintes
sélectives qui opèrent sur ces séquences:
- étudier des données de transcriptome (chez l'homme) pour quantifier l'efficacité des signaux
d'épissage dans différents tissus
- analyser les corrélations entre efficacité d'épissage et différents paramètres (séquence des
signaux d'épissage, composition en base des introns, taille des introns, niveau d'expression
des gènes, …)
- quantifier la pression de sélection sur chaque position des signaux d'épissage par analyse des
données de polymorphisme (projet 1000 génomes humains)
Ce projet implique d'analyser de gros volumes de données (RNAseq, polymorphisme). L'étudiant
devra développer des scripts pour gérer et analyser les données, mettre en œuvre des outils
bioinformatiques d'analyse de transcriptomes, et des outils statistiques d'analyse de données (R).
L'étudiant utilisera également les concepts de la génomique comparative pour déterminer l'intensité
des contraintes fonctionnelles opérant sur les séquences.
1
/
1
100%