Le syndrome de Marfan - John Libbey Eurotext

Maladies rares
Sang Thrombose Vaisseaux 2003 ;
15, n° 2 : 110–5
Le syndrome de Marfan
Guillaume Jondeau, Catherine Boileau, Bertrand Chevallier, Gabriel Delorme, Magdalena de Saint-Jean,
Franck Digne, Chabnam Guiti, Bertrand Moura, Christine Muti
Consultation Multidisciplinaire Marfan, Hôpital Ambroise-Paré, 9, avenue Charles-de-Gaulle, 92100 Boulogne
Correspondance :
G. Jondeau
Mots clés : anomalie de la
fibrilline, syndrome de Marfan,
dissection de l’aorte
L’essentiel de la maladie
La maladie de Marfan est une maladie génétique, de transmission dominante
autosomique, en rapport avec une anomalie de la fibrilline. Ses traductions
multiples témoignent d’une faiblesse du tissu de soutien et notamment de la
paroi aortique : dilatation et dissection aortiques sont responsables de la
surmortalité associée au syndrome. Le diagnostic est parfois délicat du fait de
la multiplicité des systèmes atteints (ophtalmologique, rhumatologique, cu-
tané, pulmonaire, neurologique et cardiologique), et nécessite la confronta-
tion de l’avis de différents spécialistes qui recherchent les critères diagnosti-
ques admis. Le traitement bêtabloquant limite la dilatation aortique, la
surveillance échocardiographique régulière permet de proposer la chirurgie
de remplacement de l’aorte ascendante avant la dissection.
Introduction
Depuis la description en 1896 du cas d’une jeune fille présentant une dolichos-
ténomélie par Marfan [1], la compréhension de ce syndrome a beaucoup évolué.
Le syndrome de Marfan, associe un ensemble de signes cliniques squelettiques,
oculaires, cutanés, pulmonaires et cardio-vasculaires. Des personnages célèbres
l’ont présentée et certains ont tiré partie de l’agilité qu’il leur conférait : Nicolo
Paganini, Serge Rachmaninov, Franklin Roosevelt sont suspects d’avoir pré-
senté ce syndrome (figure 1). Les progrès médicaux ont d’abord concerné
l’appréciation de son retentissement cardiovasculaire, responsable de la sur-
mortalité associée au syndrome, puis la mise en évidence des mutations respon-
sables du syndrome.
Le syndrome de Marfan est très généralement la conséquence d’une mutation
du gène de la fibrilline de type I [2], et se transmet selon le mode autosomique
dominant. Sa fréquence est difficile à apprécier du fait de la méconnaissance
probable de nombreux cas et a été estimée à 3-5/10 000, sans prédominance de
race ou sexe. Sa pénétrance est très élevée, c’est-à-dire que le porteur de
l’anomalie génétique présente presque toujours le syndrome après un certain
âge, si bien que l’un des parents d’un enfant atteint est atteint, sauf s’il s’agit
d’une nouvelle mutation, ce qui serait le cas dans un tiers à un quart des cas, et
peut-être plus si le patient présente une forme sévère.
STV, n° 2, vol. 15, février 2003
110
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 05/06/2017.

Critères
Les critères diagnotiques du syndrome de Marfan ont beau-
coup évolué au cours du temps. Au point que la jeune fille
que Marfan a décrit initialement présentait probablement
une arachnodactylie contracturante congénitale et non un
syndrome de Marfan. Ceci illustre les difficultés diagnosti-
ques parfois rencontrées. Des critères ont été proposés [3],
et récemment, des arguments génétiques ont été introduits
[4]. Ces tentatives de standardisation n’ont pas résolu tous
les problèmes.
Manifestations cliniques
Atteinte squelettique
La taille est typiquement élevée. Chez les enfants comme
chez les adultes elle dépasse le 97 ° percentile pour le sexe
et l’age. Néanmoins, la taille est rarement celle de vrais
géants : elle est généralement inférieure à 1,95 m chez
l’homme et 1,85 m chez la femme [5].
L’envergure, c’est-à-dire la distance entre l’extrémité des
doigts des deux mains, lorsque les bras sont écartés de la
poitrine à l’horizontale, est souvent supérieure à 1,05 fois la
taille. Les bras sont longs et les jambes sont grandes (doli-
chosténomélie), ce que l’on quantifie en calculant le rapport
segment supérieur (sommet du crâne-symphyse
pubienne)/segment inférieur (symphyse pubienne-sol). Ce
rapport est particulièrement abaissé chez les sujets atteints
par rapport aux sujets normaux mais la valeur normale
dépend de l’âge, du sexe et de la race. Enfin, comme les os
longs sont d’autant plus anormalement longs qu’ils sont
situés en périphérie, les manifestations sont maximales au
niveau des mains, réalisant l’aspect de « main en araignée »
(arachnodactylie) lorsque les doigts à demi fléchis reposent
sur un plan dur par leur extrémité. Au niveau des mains,
l’existence de longs doigts et d’un poignet fin est mise en
évidence par le signe du poignet : le sujet en enserrant son
Figure 1.Quelques « Marfan présumés » célèbres.
STV, n° 2, vol. 15, février 2003 111
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 05/06/2017.

poignet avec l’autre main peut atteindre et même couvrir le
pouce avec l’auriculaire.
La déformation thoracique est la conséquence d’une crois-
sance excessive des côtes, ce qui peut entraîner pectus
excavatum ou pectus carinatum. La colonne vertébrale peut
être le siège d’une cyphoscoliose, elle aussi parfois chirur-
gicale, et/ou d’un dos plat. Ces déformations se dévelop-
pent surtout au cours de la puberté et peuvent lorsqu’elles
sont très prononcées justifier un traitement chirurgical.
L’hyperlaxité ligamentaire peut être responsable d’une ins-
tabilité des articulations. Le pied plat est une complication
classique et fréquente. Au niveau de la main, le « signe du
pouce » ou signe de Steinberg est caractéristique : lorsque
le pouce est placé en opposition avec la paume, son extré-
mité dépasse le bord cubital de la main.
Atteinte oculaire
Le globe oculaire est souvent allongé, ce qui contribue au
développement d’une myopie, elle-même facteur de risque
d’un décollement rétinien. La cornée est plus plate que
normalement et la dilatation de la pupille plus difficile à
obtenir avec les mydriatiques usuels en raison de l’hypopla-
sie du muscle pupillaire. Le cristallin est souvent déplacé
du fait de la faiblesse des ligaments oculaires : lorsque le
cristallin reste tenu par les ligaments oculaires, mais qu’il
est déplacé on parle d’ectopie du cristallin. On parle de
luxation lorsqu’il a perdu toute attache avec les ligaments,
et le déplacement du cristallin se fait alors soit en avant, soit
en arrière. La sub-luxation du cristallin peut conduire à
l’ectopie cristalline. La cataracte survient plus rapidement
chez les patients atteints d’un syndrome de Marfan.
Autres atteintes
Peau
Les vergetures sont fréquentes, aux endroits où l’élasticité
de la peau est particulièrement sollicitée, comme la région
lombaire, les épaules, les seins... Elles résultent de la disso-
ciation des fibres élastiques, visible en histologie. Les her-
nies sont plus fréquentes chez les patients atteints de syn-
drome de Marfan que dans la population générale et
récidivent fréquemment après chirurgie, nécessitant sou-
vent l’utilisation de plaques.
Canal rachidien
Le sac dural se distend du fait de la pression du liquide
céphalorachidien, ce qui peut entraîner une dilatation du
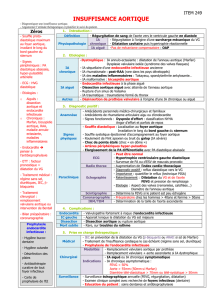
Tableau 1. Signes à rechercher
pour le diagnostic de Marfan [4].
(Les signes majeurs sont en caractère gras ;
pour parler d'une atteinte d'un système,
il faut qu'un nombre minimal de signes soit présent).
1) Squelettiques (>4 signes en gras pour un signe majeur
squelettique)
Pectus carinatum, ou excavatum
nécessitant la chirurgie
Rapport segment supérieur sur segment inférieur bas,
ou envergure sur taille > 1,05
Signe du poignet ou du pouce
Scoliose > 20 ° ou spondylolisthesis
Extension maximale des coudes < 170 °
Pied plat
Protrusion acétabulaire
Pectus excavatum modéré
Hyperlaxité ligamentaire
Palais ogival avec chevauchement des dents
Faciès
2) Oculaires (>2 signes mineurs pour une atteinte oculaire)
Ectopie cristalline
Cornée plate
Globe oculaire allongé
Iris hypoplasique ou hypoplaie du muscle ciliaire
3) Cardiovasculaires (>1 signe mineur pour une atteinte
cardiaque)
Dilatation de l’aorte ascendante intéressant les sinus
de Valsalva
Dissection aortique
Insuffisance aortique
Prolapsus valvulaire mitral avec ou sans fuite
Dilatation de l’artère pulmonaire avant l’age de 40 ans
Calcifications de l’anneau mitral avant l’age de 40 ans
Anévrisme ou dissection de l’aorte abdominale avant l’age de
50 ans
4) Pulmonaires (>1 signe mineur pour une atteinte pulmonaire)
Pneumothorax spontané
Bulle apicale
5) Cutanés (>1 signe mineur pour une atteinte cutanée)
Vergetures (à l’exclusion de : grossesse, perte de poids)
Hernies récidivantes
6) Dure mère
Ectasie de la dure mère lombo-sacrée
7) Génétique
Un parent direct ayant les critères diagnostics
Mutation de FBN 1 déjà connue pour provoquer un
syndrome de Marfan
Présence d’un marqueur génétique, proche du gène
de la fibrilline de type I, se transmettant avec la
maladie dans la famille.
STV, n° 2, vol. 15, février 2003
112
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 05/06/2017.

canal lombaire. Au maximum, le canal vertébral peut être le
siège d’une véritable hernie avec méningocèle. Clinique-
ment, cette anomalie est le plus souvent asymptomatique,
mais peut entraîner une compression, généralement radicu-
laire. Elle est facilement mise en évidence par la tomoden-
sitométrie ou l’IRM, et peut être recherchée par une radio-
graphie de rachis lombaire de profil.
Poumons
La complication pulmonaire la plus classique est le pneu-
mothorax spontané, par rupture d’une bulle apicale. Les
anomalies observées lors des épreuves fonctionnelles res-
piratoires sont la conséquence des déformations thoraci-
ques ou le résultat de la surestimation des capacités théori-
ques du fait de la longueur excessive des jambes hors de
proportion avec le développement thoracique.
Manifestations cardiovasculaires
Son atteinte centrait le pronostic vital avant que la chirurgie
de remplacement de l’aorte ascendante ne se soit dévelop-
pée : 90 % des patients présentant un syndrome de Marfan
décédaient alors de complications cardiovasculaires, dis-
section et rupture aortique, insuffisance aortique ou mitrale
ou l’insuffisance cardiaque qui peut en résulter [6]. Depuis
l’amélioration de la prise en charge, le pronostic des pa-
tients s’est transformé et l’espérance de vie était de 72 ans
sur une étude publiée en 1995 [7]. Il est utile de faire le
diagnostic !
Atteinte valvulaire
La valve mitrale est souvent redondante, avec allongement
des cordages et dilatation de l’anneau mitral, comme dans
la maladie de Barlow. Il peut en résulter une insuffisance
mitrale parfois importante et il s’agit de la principale com-
plication cardiaque chez les patients jeunes. Mais la fuite
valvulaire peut aussi se développer avec le temps.
La grande majorité des fuites valvulaires aortiques résulte
de la dilatation de l’aorte ascendante, au niveau de la partie
supérieure de l’attache valvulaire, au-dessus de l’anneau
aortique proprement dit qui n’est habituellement pas dilaté,
au moins au début. Il est donc au moins théoriquement
possible de restaurer la continence de l’orifice en reformant
la géométrie de la racine aortique.
Anomalies de l’aorte
La paroi aortique des patients présentant un syndrome de
Marfan est fragilisée : son élasticité est altérée, et ce sur
l’ensemble de son trajet, thoracique et abdominal. Mais la
propriété élastique des parois des autres vaisseaux n’est pas
modifiée [8]. En conséquence, l’aorte initiale se dilate,
typiquement en « bulbe d’oignon ». Cette dilatation peut
débuter in utero et sa progression est très difficile à prévoir.
Deux complications sont à craindre : tout d’abord une fuite
aortique peut résulter de la modification de la position des
feuillets valvulaires (du fait de la dilatation), et dépend
donc directement du degré de dilatation de l’aorte initiale.
Ensuite et surtout, le risque de dissection ou de rupture
aortique.
Bien que la dilatation et le risque de dissection prédominent
au niveau de l’aorte ascendante, ces complications peuvent
également survenir sur l’aorte thoracique descendante ou
abdominale (les propriétés élastiques de la paroi aortique
sont modifiées sur l’ensemble de son trajet). Ceci est impor-
tant à considérer pour la thérapeutique (et il faudra poursui-
vre les béta-bloquants même après remplacement de l’aorte
ascendante) et pour la surveillance (il faut surveiller l’aorte
dans son ensemble).
Critères pronostiques :
facteurs favorisant la dissection aortique
La dissection de l’aorte ascendante a d’autant plus de risque
de survenir que
•Le diamètre aortique est plus élevé ; on considère que le
risque est faible (bien que non nul) lorsque le diamètre
aortique au niveau des sinus de Valsalva reste en dessous de
50 à 55 mm.
•Le diamètre aortique augmente, en valeur absolue chez
l’adulte ou plus que ne le voudrait la croissance chez
l’enfant : la dilatation rapide d’une aorte jusque là stable
doit faire considérer l’intervention avant que la dissection
ne survienne.
•Il existe une histoire familiale de dissection de l’aorte :
bien qu’une grande variabilité phénotypique soit observée à
l’intérieur d’une même famille, il semble que le risque de
dissection précoce soit plus important si un autre membre
de la famille a présenté une dissection aortique jeune ou
alors que les sinus de Valsalva n’étaient que peu dilatés.
•Le patient réalise des efforts isométriques qui s’accom-
pagnent d’une augmentation importante de la pression arté-
rielle systolique et augmente ainsi la contrainte appliquée à
l’aorte initiale. Il faut donc déconseiller les sports qui
impliquent ce type d’effort, tels le basket ball, le tennis, le
hand-ball, le volley-ball... et bien sur la musculation que ces
patients pourraient être désireux de la pratiquer du fait de la
diminution de la masse musculaire qui accompagne parfois
le syndrome.
STV, n° 2, vol. 15, février 2003 113
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 05/06/2017.

•Le patient ne prend pas de traitement bêta-bloquant (voir
ci-après)
•Qu’une femme est enceinte (voir ci-après)
•Que le patient est hypertendu.
Principes thérapeutiques
Possibilités thérapeutiques cardiovasculaires
La prévention de la dissection aortique repose sur la limita-
tion des activités physiques, le traitement bêta-bloquant et
la chirurgie préventive de remplacement de l’aorte ascen-
dante.
La limitation de l’activité physique dépend du risque
supposé de dissection aortique : une dilatation modérée de
l’aorte ascendante ne justifie pas d’interdire toute activité
sportive. Il faut naturellement éviter les sports de combats,
les sports en compétition surtout s’ils comportent des ef-
forts en apnée ou des risques de collision. La restriction de
l’activité physique pourrait aussi limiter la rapidité de la
dilatation aortique.
Le traitement pharmacologique repose sur la prise au long
cours de b-bloquants : en diminuant la pression artérielle et
sa vitesse d’ascension (dP/dt), ils ralentissent la progres-
sion de la dilatation aortique, et ce indépendamment du
diamètre aortique au moment de l’institution du traitement.
Enfin, ils diminuent le taux de complications aortiques.
Leur bénéfice clinique a été démontré chez les patients de
plus de 12 ans [9]. L’attitude à adopter chez les enfants est
clairement définie, et l’effet en cas de dilatation aortique
majeure est moins évident. Il faut les poursuivre après
chirurgie.
La chirurgie cardiaque préventive peut être indiquée pour
corriger une fuite valvulaire (mitrale ou aortique), en cas de
dilatation aortique importante ou de dissection aortique
chronique. Au niveau de la valve mitrale, la redondance des
feuillets mitraux et la dilatation de l’anneau mitral souvent
observées permettent, comme dans le prolapsus mitral, de
faire une chirurgie réparatrice selon la technique de Car-
pentier et l’expérience montre que son efficacité est dura-
ble.
La chirurgie aortique repose essentiellement sur la répara-
tion de l’aorte ascendante avec remplacement valvulaire
aortique (opération de Bentall) ou non (intervention de T
David ou de Yacoub). Le risque d’une telle opération
lorsqu’elle est effectuée à froid est très faible et son déve-
loppement a permis d’augmenter l’espérance de vie des
patients [10]. La tendance est d’opérer de plus en plus tôt :
la valeur de 55 mm est généralement acceptée [11], et la
valeur de 50 mm de plus en plus souvent proposée. En fait,
il importe de prendre en compte non seulement la valeur
absolue du diamètre aortique mesuré mais aussi son évolu-
tion au cours du temps, paramètre qui est de plus en plus
souvent disponible grâce à la surveillance régulière des
patients par échocardiographie. Il est de règle d’effectuer
une échocardiographie annuelle pour surveiller le diamètre
de l’aorte ascendante, particulièrement au niveau des sinus
de Valsalva. La technique de mesure doit être standardisée
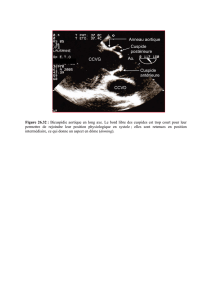
(figure 2).
Les indications chirurgicales en cas de dissection aortique
sont identiques à celles qui sont préconisées dans les dis-
sections classiques, mais le remplacement valvulaire aorti-
que associé est fait de façon quasi-constante en cas d’at-
teinte de l’aorte ascendante.
Figure 2.Technique de mesure échocardiographique du diamètre
aortique.
STV, n° 2, vol. 15, février 2003
114
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 05/06/2017.
 6
6
1
/
6
100%