A Young Adult with Aplastic Anemia and Gray

A Young Adult with Aplastic Anemia and
Gray Hair
Jeune adulte atteint d'anémie aplasique aux
cheveux gris
Eva C. Guinan
1
,
2
,
3
,
4
,* et Suneet Agarwal
2
,
3
,
4
,
5
Affiliations des auteurs
1
Departments of Radiation Oncology and
2
Pediatric Oncology, Dana-Farber Cancer Institute, Boston, MA;
3
Division of Hematology/Oncology, Boston Children's Hospital, Boston, MA;
4
Harvard Medical School, Boston, MA;
5
Harvard Stem Cell Institute, Harvard University, Cambridge, MA.
* Adresser toute correspondance à cet auteur à : Dana-Farber Cancer Institute, 450 Brookline
Ave., Boston, MA 02215. Fax 617-632-3770; e-mail [email protected].
DESCRIPTION DU CAS
Un homme âgé d'une vingtaine d'années s'est présenté avec une hématurie et une pancytopénie
modérée [leucocytes, 1,9 × 10
9
/l (référence, 4,2 × 10
9
/l à 9,9 × 10
9
/l) ; hémoglobine, 13,3 g/dl
(référence, 13,0 à 17,4 g/dl) ; plaquettes, 61 × 10
9
/l (référence, 140 × 10
9
/l à 440 × 10
9
/l)].
L'examen physique était normal, ainsi que les tests de laboratoire de routine. Un aspira de moelle
osseuse (MO)
6
et une biopsie ont démontré une hypo-cellularité (20 %) sans dysplasie. Les
résultats de la cytogénétique de la MO, de l'hybridation fluorescente in situ (FISH), et d'un test
au diépoxybutane pour l'anémie de Fanconi étaient normaux. Les résultats de la cytométrie en
flux pour le sang périphérique n'ont mis en évidence aucun trouble lymphoprolifératif ni de
maturation myéloïde, ni aucune anomalie de protéine cohérente avec une hémoglobinurie
paroxystique nocturne. Au cours des 5 années qui ont suivi, le patient est demeuré
asymptomatique, n'a reçu aucune transfusion tout en présentant une numération globulaire stable
et une histologie de MO inchangée.
Dans le cadre d'une étude portant sur des interventions thérapeutiques potentielles, le patient a
recherché un second avis, et il s'est alors souvenu d'avoir souffert de sténoses urétrales
récurrentes à l'âge de 6 à 9 ans et à nouveau à 22 ans. Ses cheveux ont commencé à blanchir à 11
ans, et son front s'est dégarni dès l'âge de 16 ans. Il existe dans la famille des antécédents de
constatations similaires. Il a décrit ses ongles des mains et des pieds comme toujours
"terriblement" secs et craquelés. Il a rapporté souffrir d'un excès de larmes et que ses amis

avaient remarqué qu'il pleurait alors que lui-même n'était pas conscient de la production de
larmes. À l'examen, le patient présentait des cheveux clairsemés, grisonnants et une calvitie
médio-frontale. Il affichait une pigmentation réticulaire brune légèrement rougeâtre, sans
blancheur, subtile sur la partie supérieure antérieure et postérieure de son thorax. Tous ses ongles
étaient nettement dystrophiques. Une lésion blanche plate de 1 × 1,5 cm était présente sur sa
voûte palatine. Le reste de l'examen était normal. Ses numérations sanguines avaient été plutôt
stables au cours des 5 années précédentes : leucocytes, 1,4 × 10
9
/l à 3,8 × 10
9
/l (référence, 4,2 ×
10
9
/l à 9,9 × 10
9
/l) ; nombre absolu des neutrophiles, 0,71 × 10
9
/l à 2,7 × 10
9
/l (référence, 2,4 ×
10
9
/l à 7,6 × 10
9
/l) ; hémoglobine, 12,9 à 14,5 g/dl (référence, 13,0 à 17,4 g/dl), avec une
macrocytose marquée (volume globulaire moyen, 105 à 111 µ
3
; référence, 82,0 à 100 µ
3
) ; et
plaquettes, 46 × 10
9
/l à 71 × 10
9
/l (référence, 140 × 10
9
/l à 440 × 10
9
/l).
QUESTIONS A PRENDRE EN CONSIDERATION
1. Quelles sont les causes de l'anémie aplasique (AA) ?
2. Étant donné les symptômes du patient, quel pourrait être le diagnostic ?
3. Quelles analyses de laboratoire seraient utiles pour le diagnostic ?
DISCUSSION
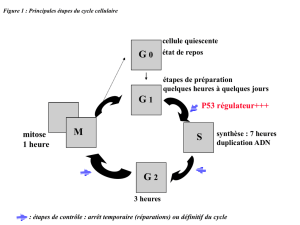
Le patient s'est présenté comme un adulte atteint d'une AA modérée, définie par une MO
hypocellulaire avec diminution des numérations globulaires dans le sang périphérique dans plus
d’une lignée cellulaire. La plupart des cas d'AA se classent idiopathiques ou acquis et il est le
plus souvent supposé qu'ils sont dus à une dérégulation immunitaire, peut-être à cause d'une
infection virale. Des expositions à une dose substantielle de radiation, certains médicaments, et
produits chimiques, ainsi que l'apparition de certains syndromes auto-immuns, ont été également
associés à une insuffisance médullaire. Des affections hématologiques, y compris une
myélodysplasie, une hémoglobinurie paroxystique nocturne, et, rarement, une leucémie peuvent
également se présenter sous la forme d'une AA. En variante, une évaluation soigneuse de
l'historique du patient, un examen physique, et les antécédents familiaux peuvent suggérer un
syndrome héréditaire d'insuffisance de la MO (IBMFS) comme étiologie. Ce patient a affiché
plusieurs caractères de dyskératose congénitale (DKC) sous sa forme classique (dystrophie des
ongles, leucoplasie mucosale, et variations de la pigmentation cutanée) et de concert avec
d'autres anomalies, y compris une insuffisance de la MO, des sténoses urétrales, une production
excessive de larmes (épiphora), une calvitie et des cheveux blancs prématurés, ainsi qu'une
pneumopathie (1).
SUIVI DU PATIENT
Sur la base d'un diagnostic clinique présomptif de DKC, nous avons entrepris des mesures de la
longueur des télomères (LT), qui nous ont montré qu'à la fois ses lymphocytes et ses
granulocytes présentaient une LT médiane (LTM) réduite de façon marquée (<1er percentile)
[4,1 kb contre 7,5 kb (50ème percentile pour la tranche d'âge) et 4,5 kb contre 8,6 kb (50ème
percentile pour la tranche d'âge), respectivement], confirmant de cette manière le diagnostic
(Fig. 1). Nous avons également entrepris des analyses à la recherche de mutations dans des gènes

connus comme étant associés à une DKC, y compris le séquençage de DKC1
7
(dyskératose
congénitale 1, dyskérine), de l'exon 6 de TINF2 [facteur nucléaire 2 interagissant avec TERF1
(TRF1)], et de TERC (composant ARN de la télomérase). Nous avons également entrepris une
analyse des délétions/duplications du gène TERC. Il n'a été identifié aucune anomalie. Les
résultats de la densitométrie osseuse (absorptiométrie bi-énergétique aux rayons X) étaient
normaux. L'analyse de la fonction pulmonaire a révélé une valeur de capacité de diffusion du
monoxyde de carbone du poumon (ou DLCO) de 17,0 ml · mmHg
−1
· min
−1
(45 % de la valeur
prédite). La biopsie de la lésion de la voûte palatine a mis en évidence une dysplasie épithéliale
légère et une hyperkératose.
L'identification des mutations génétiques sous-jacentes des IBMFS ces 20 dernières années a
révélé une variation marquée dans l'apparition et les manifestations cliniques provoquées par des
mutations similaires. Ainsi, à la fois pour les enfants et les adultes, les hématologues doivent
maintenir un haut niveau de suspicion pour des causes génétiques lors de l'évaluation de patients
atteints d'insuffisance de la MO. De nombreux modes de transmission (autosomique dominante
et récessive, liée au chromosome X, sporadique), un taux variable de pénétrance, une pléiotropie,
et une anticipation génétique compliquent le diagnostic de la DKC (2). Les patients atteints de
troubles liés à une DKC peuvent afficher un spectre clinique englobant une pathologie
multisystémique sévère dès l'enfance (par exemple, les syndromes de Hoyeraal–Hreidarsson et
Revesz) et d’apparition tardive, des troubles à restrictions tissulaires comme la mucoviscidose.
Des mutations dans l'un des 8 gènes impliqués dans la biologie des télomères—DKC1, TINF2,
TERC, TERT (transcriptase inverse de la télomérase), NHP2 [homologue de la
ribonucléoprotéine NHP2 (levures)], NOP10 [homologue de la ribonucléoprotéine NOP10
(levures)], WRAP53 (contenant des répétitions WD, antisens par rapport à TP53 ; anciennement
TCAB1), et CTC1 (composant 1 du complexe de maintenance des télomères CTS)— peuvent être
identifiées chez approximativement 50 % des patients présentant des caractères cliniques de
DKC classique, le restant étant encore non caractérisé (3, 4). Les télomères se situent aux
extrémités des chromosomes et sont constitués de plusieurs centaines à plusieurs milliers de
répétitions d'un hexanucléotide [(TTAGGG)
n
], qui subissent une attrition avec chaque division
cellulaire. Des complexes de protéines multiples (par exemple, incluant des composants codés
par TINF2 et CTC1) maintiennent les télomères, et une ribonucléoprotéine, la télomérase
[composée des produits des gènes TERC, TERT, DKC1, TCAB1 (c'est-à-dire, WRAP53), NHP2,
et NOP10] remplace les répétitions hexanucléotidiques par transcription inverse pour
contrecarrer l'attrition télomérique. Les lésions génétiques dans la DKC compromettent l'intégrité
des télomères, altérant la capacité d'auto-renouvellement et régénérative dans les cellules, et
expliquent éventuellement l'insuffisance prématurée de multiples organes chez les individus
touchés (5).
Une maintenance défectueuse des télomères est un caractère courant parmi les génotypes de la
DKC, qui peut être exploité pour le diagnostic. Lansdorp et ses collègues ont développé une
approche par une technique FISH basée sur la cytométrie en flux (FISH en flux) certifiée par la
CLIA pour quantifier les LT dans les cellules du sang périphérique (6). Des sondes d'acides
nucléiques peptidiques marquées par fluorescence (CCCTAA)
3
sont hybridées à des télomères
dans des cellules perméabilisées du sang périphérique. Le nombre des sondes liées aux télomères
est proportionnel à la LT, et l'intensité de fluorescence totale des sondes liées aux 92 extrémités
télomériques des 46 chromosomes est interprétée comme la LTM par cellule. Du fait que les

télomères se raccourcissent de façon normale dans les cellules sanguines avec l'âge, Lansdorp et
al. ont établi des normes ajustées à l'âge pour la LTM mesurée par la technique FISH en flux.
Une coloration simultanée avec des anticorps pour les marqueurs de la surface cellulaire permet
une mesure de la LTM dans des sous-ensembles spécifiques des cellules du sang périphérique
(par exemple, les lymphocytes, les granulocytes). La comparaison d'un échantillon du patient
avec des témoins en bonne santé permet d'attribuer un percentile normalisé pour l'âge et le type
cellulaire à la LTM du patient.
Alter et al. ont utilisé la technique FISH en flux pour mesurer la LTM dans les sous-ensembles
des cellules du sang périphérique de 65 patients atteints de DKC (définis comme affichant des
anomalies cliniques classiques ou porteurs d'une mutation pathogène connue) contre 127
membres de familles cliniquement en bonne santé (7). En utilisant des rangs de LTM <1er
percentile (“très bas”) comme valeur seuil, Alter et al. ont montré que la FISH en flux offre une
sensibilité et une spécificité pour le diagnostic de la DKC, respectivement, de 97 % et 91 %, avec
les lymphocytes totaux, et de 97 % et 82 % avec les granulocytes. La sensibilité et la spécificité
pour le diagnostic de la DKC ont été étudiées avec des sous-ensembles de lymphocytes, mais les
meilleures performances de la technique FISH en flux ont été obtenues pour une seule lignée à
partir de la mesure de la LTM dans les lymphocytes totaux. Ces études importantes ont validé la
technique FISH en flux comme un test d'utilité diagnostique pour la DKC, et nous avons donc
utilisé ce test pour confirmer le diagnostic clinique d'une DKC chez ce patient (Fig. 1).
Le diagnostic génétique définitif d'une DKC demeure limité par le besoin de séquencer un
ensemble important, bien qu'incomplet, de gènes et par la difficulté à établir la pathogénicité de
séquences variables. Il a été trouvé des mutations de DKC1 dans 25 % à 30 % des cas de DKC.
DKC1 est lié au chromosome X, et les hommes sont donc touchés, tandis que les femmes
porteuses sont généralement cliniquement silencieuses. Des mutations de TINF2 sont
responsables d'approximativement 15 % des cas de DKC, elles sont autosomiques dominantes et
fréquemment sporadiques. Des mutations de TERC et TERT (pour chacun, 5 % à 10 % de tous
les cas) sont trouvées dans une DKC autosomique dominante et récessive et elles permettent
d'anticiper la maladie, de telle manière que les générations successives non seulement héritent de
la lésion génétique mais présentent aussi un raccourcissement des télomères, ce qui peut être à
l'origine d'une apparition plus précoce des manifestations cliniques (8). Une réversion somatique
du gène TERC, certainement due à une croissance sélective favorisée des cellules souches
hématopoïétiques, a été observée dans la DKC et représente un facteur de détection potentiel
dans l'analyse génétique (9). Dans un souci de rigueur clinique suffisante, un résultat normal
d'une analyse de TERC de l'ADN du sang périphérique devra être suivi de l'analyse d'autres
cellules somatiques. L'historique de notre patient était insuffisant pour déterminer le mode de
transmission, et une combinaison d'analyses de laboratoire et certifiées par la CLIA a exclu des
mutations dans les gènes DKC1, TINF2, TERC et TERT. La détermination de la LTM du patient
et de membres de sa famille par la technique FISH en flux pourrait clarifier le mode de
transmission, et le séquençage d'autres gènes candidats et de l'exome pourrait déterminer la base
génétique de la DKC du patient.
L'établissement du diagnostic d'un IBMFS contre une AA acquise présente des applications
thérapeutiques significatives. Comme dans ce cas, des manifestations retardées et bénignes
doivent être prises en considération chez des adultes souffrant d'une insuffisance de la MO.

L'insuffisance de la MO de l'IBMFS répond en général médiocrement à la cyclosporine A et à un
traitement immunosuppresseur à base de globulines antithymocytaires utilisé pour l'AA acquise
en l'absence de donneur compatible de la même famille pour une transplantation de cellules
hématopoïétiques (TCH). En outre, les protocoles préparatifs de la TCH utilisés pour l'AA sont
associés à une toxicité inacceptable pour les organes chez les patients atteints de DKC, alors que
des protocoles préparatifs spécifiques des pathologies plus récents d'une intensité réduite ont
produit des résultats améliorés. Lorsqu'un IBMFS est suspecté, il est impératif d'exclure une
maladie sub-clinique chez les donneurs potentiels de la même famille pour une TCH. Après une
insuffisance de la MO, les causes majeures de décès dans la DKC comprennent des cancers des
cellules squameuses de la tête, du cou, et de la zone génito-anale, une mucoviscidose, et une
cirrhose (10). Bien que la relation physiopathologique de l'intégrité défectueuse des télomères
avec ces complications demeure spéculative, la plage des manifestations potentiellement fatales
souligne la pertinence d'un criblage familial, même en l'absence de TCH envisagée. Par
conséquent, le diagnostic d'une DKC et d'autres IBMFS a non seulement un impact sur le
traitement immédiat mais mandate également des soins multidisciplinaires coordonnés, une
surveillance à long terme, des conseils de prévention, ainsi que pour la famille et le conseil
génétique.
POINTS A RETENIR
•
Une anémie aplasique peut survenir suite à une dérégulation immunitaire et à une
exposition à des agents toxiques, ainsi que comme manifestation de plusieurs troubles
hématologiques, y compris une myélodysplasie et un IBMFS.
•
Une cause génétique devra être envisagée chez des patients de tous les âges qui se
présentent avec une AA. Une évaluation de base pour une DKC devra comprendre un
examen physique méticuleux, l'historique médical, et les antécédents familiaux.
•
L'analyse génétique qui devra être envisagée dans l'évaluation de la DKC comprend la
mesure de la LT dans les cellules du sang périphérique et l'analyse de mutations
génétiques des gènes candidats.
•
Bien que la DKC soit provoquée par des lésions dans plusieurs gènes connus pour
compromettre la maintenance des télomères, les gènes responsables demeurent inconnus
dans 50 % des cas.
•
L'établissement du diagnostic d'une DKC contre une AA idiopathique représente des
implications majeures pour le patient et sa famille.
Remerciements
Nous sommes reconnaissants envers Repeat Diagnostics Inc., North Vancouver, British
Columbia, Canada, qui nous a fourni les données d'analyses de la longueur des télomères sur la
Fig. 1.
Notes
6
Abréviations non standard :
 6
7
8
9
10
11
12
13
6
7
8
9
10
11
12
13
1
/
13
100%