Cours Prepa agreg 2012

Métabolisme cellulaire - 1
LA CATALYSE ENZYMATIQUE
Introduction
La cellule peut être considérée comme un système thermodynamique ouvert. Elle est le siège de très nombreuses
réactions, soumises elles aussi aux lois thermodynamiques (réactions spontanées si ΔrG<0). Mais une réaction
peut aussi être envisagée sous l’angle de la cinétique : quelle est la vitesse de la réaction ? A quelle vitesse est
atteint l’équilibre de réaction ?
La totalité des réactions du métabolisme cellulaire sont catalysées, par des enzymes. Les enzymes toutes des
protéines. Qu’est-ce qu’un catalyseur ? Quelles sont les propriétés propres aux catalyseurs biologiques que sont
les enzymes ? En quoi leur nature de protéines leur permet de réaliser cette fonction de catalyseur ?
I. LES ENZYMES, DES CATALYSEURS BIOLOGIQUES
A. Action du suc pancréatique sur l’amidon
Une expérience simple, dans un bain marie thermostaté à 37°. On place 2 récipients :
Expérience 1
Tube 1 : solution d’empois d’amidon
Tube 2 : solution d’empois d’amidon + suc pancréatique
On prélève une partie du contenu à intervalles réguliers puis on réalise un test à l’eau iodée et un test à la liqueur
de Fehling.
Résultats
Temps en min
0
5
10
15
tube 2 eau iodée
+
+/-
+/-
-
tube 2 liqueur Fehling
-
-
-
+
tube 1 eau iodée
+
+
+
+
tube 1 liqueur Fehling
-
-
-
-
Interprétation
En présence de suc pancréatique, l’amidon est hydrolysé en un sucre réducteur
Amidon n maltose
La vitesse de la réaction est nulle dans les conditions expérimentales du tube 1.
Conclusion : les enzymes présentes dans le suc pancréatique catalysent la réaction d’hydrolyse de l’amidon. C’est-
à-dire de rupture des liaisons covalentes O glycosidiques.
3ème conditions expérimentales : bain marie à 100° + HCl concentré + amidon
On observe alors une disparition de l’amidon et une apparition de sucre réducteur (maltose)
Donc dans ces conditions l’hydrolyse a lieu, on parle de catalyse acide.
La catalyse correspond à une augmentation de la vitesse de réaction, ici les enzymes pancréatiques (amylase) et
le HCl jouent le rôle de catalyseur mais dans des conditions bien différentes.
amylase
HCl
Catalyse l’hydrolyse de l’amidon cuit
uniquement
Idem,
mais aussi d’autres réactions
Substrat = amidon seulement
Action sur d’autres polyosides : cellulose…
Action à faible concentration
Action à forte concentration
T=37°
T=100°
Quelles sont les propriétés des enzymes communes à tous les catalyseurs ?
B. Les enzymes accélèrent les réactions sans modifier l’équilibre final
Soit une réaction A + B C + D
Comme toute réaction, on peut calculer un rG. Dans le cas de la réaction considérée, rG<0 dans le sens A + B
C + D, cette réaction est exergonique. En abscisse, on suit l’avancement de la réaction. Deux paliers sont
représentés : état initial et état d’équilibre de la réaction.

Métabolisme cellulaire - 2
On voit que la réaction (comme toute réaction) nécessite le passe par un état de transition, de niveau
énergétique plus élevée. On peut considérer que c’est le passage par des intermédiaires de réactions plus
instables.
Il existe donc une sorte de barrière énergétique : il faut un apport d’énergie pour passer cette barrière, c’est
l’énergie d’activation. Même si le bilan global de la réaction est une libération d’énergie (rG<0). L’apport
d’énergie peut être réalisée par une augmentation de la température par exemple.
Les enzymes (et les catalyseurs en général) agissent en diminuant l’énergie d’activation, la conséquence est une
accélération de la réaction (augmentation de la vitesse). On verra plus tard que la formation du complexe enzyme
substrat permet de stabiliser l’état de transition et par conséquent d’abaisser l’énergie d’activation.
Par contre, on remarque que le rG reste inchangé. L’état d’équilibre n’est pas modifié, la constante d’équilibre
reste identique.
BA DC
Ke .
.
Les concentrations à l’équilibre restent inchangées.
En d’autres termes, une enzyme ne peut rendre spontanée une réaction endergonique. Un même état d’équilibre
est simplement atteint plus rapidement. Les constantes de vitesse des réactions opposées sont augmentées dans
les mêmes proportions.
Autres propriétés de catalyseurs :
- l’enzyme se retrouve intact à la fin de la réaction et en même quantité : il n’est pas consommé ou produit
par la réaction
- Par conséquent, une enzyme peut catalyser la réaction n fois selon un cycle d’action. Ce qui explique
qu’elles agissent à faible concentration.
Vocabulaire : A et B substrats de l’enzymes, C et D produits de la réaction.
C. Les conditions physico-chimiques influencent les propriétés catalytiques des enzymes
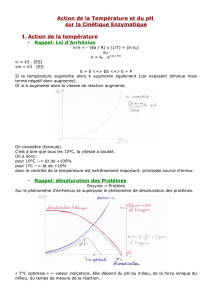
1) Effet de la température
Courbe de la vitesse en fonction de la température.
Résultats : a) entre 0 et 40 la vitesse de la réaction augmente lorsque la température augmente.
b) au-delà d’une certaine température seuil (entre 40-45°), la vitesse de la réaction diminue rapidement avec la
température
Interprétation
a) Il s’agit d’un phénomène purement physique : toute augmentation de température provoque une
augmentation de la vitesse des réactions chimiques. L’augmentation de température provoque une augmentation
de l’agitation moléculaire et par conséquent une augmentation de la probabilité de rencontre des réactifs. Dit
autrement, l’énergie d’activation est plus basse lorsque la température est plus élevée. C’est la loi d’Arrhenius,
qui peut s’exprimer sous la relation empirique suivante qui lie k (constante de vitesse d’une réaction) et T la
température :
eRT
E
Ak
R constante des gaz parfaits, T température absolue en Kelvins
E énergie d’activation, supposée constante sur un grand intervalle de température
A constante propre à la réaction.
b) Au-delà de 45° environ (valeur variable selon les enzymes), les enzymes (comme toutes les protéines) subissent
une dénaturation. Les liaisons faibles qui maintiennent la structure III (liaisons H, liaisons ioniques, forces de Van
der Walls) se détruisent. L’enzyme perd sa conformation spatiale, elle devient par conséquent inactive : la
réaction est très ralentie. Cette dénaturation est irréversible.
On remarque qu’il résulte une température optimale pour la catalyse.
Les propriétés des enzymes sont très liées à leur nature protéique.
Remarque : certains microorganismes dits thermophiles vivent à haute température, dans de l’eau proche de 80-
100°. Leurs enzymes sont adaptées avec une température optimale plus haute, une résistance très importante à
la dénaturation par la chaleur. Citons la bactérie Thermophilus aquaticus. Elle possède une polymérase de l’ADN
très utilisée en génie génétique, la Taq polymérase utilisée pour réaliser la PCR.

Métabolisme cellulaire - 3
2) Effet du pH
Exemple de l’ARNase, enzyme catalysant l’hydrolyse de l’ARN. 3 acides aminés sont importants pour la réalisation
de la réaction :
- l’histidine 12, qui doit être à l’état déprotoné
- l’histidine 119 qui doit être à l’état protoné
- la lysine 41
En effet, le mécanisme réactionnel (sans entrer dans les détails) implique que l’His 119 donne un proton, puis que
l’His 12 accepte ultérieurement un proton.
On peut construire une courbe sur lauel apparaît le % d’His 119 protonée et le % d’His 12 déprotonée.
Résultat : on voit que la coexistence His protonée et d’His 12 déprotonée n’existe que pour un intervalle de pH
étroit légèrement supérieur à 7. Intervalle qui correspond au maximum d’activité de l’enzyme.
Interprétation : il existe un pH optimal pour l’activité enzymatique.
En effet, le pH modifie :
- au niveau de l’enzyme, l’état d’ionisation des radicaux des acides aminés (au niveau du site actif) ce qui
modifie les possibilités d’interaction avec le substrat. Le pH peut perturber la structure tridimensionnelle
(par rupture de liaison faible, hydrogène, ionique)
- au niveau du substrat, l’état d’ionisation. Ce qui modifie les possibilités d’interaction avec l’enzyme.
Pour chaque enzyme, il existe alors un pH optimal :
- souvent proche de 7,4 ; pH cellulaire comme pour l’ARNase, l’amylase salivaire.
- Mais aussi pH 2 pour la pepsine dans l’estomac ou les hydrolases lysosomiales.
Ou encore pH 8/9 pour la trypsine agissant dans l’intestin.
D. Les enzymes sont caractérisés par une spécificité de substrat et de réaction : mise en
évidence
On a observé que l’amylase contenue dans le suc pancréatique catalyse l’hydrolyse de l’amidon, mais n’agit pas
sur la cellulose.
La ribonucléase vue dans la partie précédente catalyse l’hydrolyse de l’ARN (et pas de l’ADN).
Considérons un 3ème exemple : la glucokinase, catalyse la phosphorylation du glucose dans le cytosol.
Glucose + ATP glucose6P + ADP
On a construit la courbe d’apparition du produit (glucose 6P) en fonction du temps, pour une concentration
d’enzyme et une concentration de substrat donné.
On voit que la vitesse de réaction est nulle ou presque pour tout autre substrat que le D glucose.
Les enzymes sont caractérisées par une spécificité de substrat. Elles ne peuvent agir que sur un substrat. Il s’agit
même d’une stéréospécificité (exple la glucokinase agit sur le D glucose et pas le L glucose) : il existe en fait une
complémentarité de la forme entre enzyme et substrat, à l’origine de la spécificité. Modèle clé serrure.
La spécificité vis-à-vis du substrat est plus ou moins stricte :
La glucokinase catalyse la phosphorylation du D glucose uniquement, l’hexokinase catalyse la phosphorylation des
hexoses.

Métabolisme cellulaire - 4
Les protéases, lipases, ribonucléases par exemple catalysent l’hydrolyse respectivement de protéines, de lipides,
d’ARN…
Mais il existe aussi une spécificité de réaction : les protéases catalysent l’hydrolyse de protéines, (i.e. de liaison
peptidique), les kinases catalysent des phosphorylations (ajout d’une groupement phosphate)…
La nomenclature des enzymes repose sur cette double spécificité puisque en générale le nom d’un enzyme
indique son substrat et le type de réaction catalysée : glucokinase, ribonucléase… ou non (trypsine…)
Toutefois, nous n’avons guère été précis sur l’origine de cette double spécificité, précisons maintenant quel est le
support moléculaire de cette propriété. Voyons comment encore une fois la structure protéique explique les
propriétés des enzymes. Nous répondrons alors à la questions qu’est-ce qu’un site actif.
II. LES ENZYMES, MECANISMES D’ACTION
A. Les enzymes sont caractérisées par une double spécificité : le site actif des enzymes
Les enzymes sont des protéines importance de la structure III voire IV de la protéine
1) Mutagenèse dirigée et vitesse de la réaction
Principe très général : La mutagenèse dirigée consiste à modifier la séquence du gène d’une enzyme sur un
nucléotide bien précis. On peut ensuite produire grâce à ce gène modifié, une enzyme dont un acide aminé bien
précis a été modifié. On effectue ensuite des tests d’activité de l’enzyme en mesurant la vitesse de la réaction.
Exemple ARNase déjà vue précédemment
acide aminé modifé
conséquence
His 12
V nulle
His 119
V nulle
Lys 41
V nulle
Lys 61
V peu modifiée
On observe donc que tous les acides aminés n’ont pas la même importance fonctionnelle : certains acides aminés
se regroupent dans la structure III, alors qu’ils sont éventuellement éloignés dans la structure I. Ils forment le site
actif de l’enzyme : lieu de fixation du substrat et lieu de la catalyse.
Il est possible de réaliser aussi des études cinétiques, en mesurant la vitesse initiale de la réaction en fonction de
la concentration en substrat. Voir TP pour les modalités d’obtention de la courbe.
On remarque qu’au-delà d’une certaine concentration en Substrat, la vitesse initiale reste constante (elle tend
vers Vmax). Il y a un effet de saturation. Toutes les enzymes présentes dans le milieu sont liées à une molécule de
substrat. Il existe un site de fixation du substrat, si ceux-ci sont trop nombreux, l’enzyme est saturée. Dis
autrement, il y a formation d’un complexe enzyme - substrat. La cristallographie aux rayons X confirme la
formation d’un complexe enzyme substrat au niveau d’un site spécifique de l’enzyme.
2) Notion de site actif
Le site actif de l’enzyme est le site où se fixe le substrat et où a lieu la catalyse. On distingue de façon un peu
artificielle : un site de fixation (1), un site de catalyse (2).
Le site actif correspond à un volume très restreint de l’enzyme. C’est-à-dire qu’il est constitué par un nombre
limité d’acides aminés. (cf. mutagenèse)
Le site actif est délimité par la structure 3D (structure III) de l’enzyme, c’est-à-dire le rapprochement d’acides
aminés éloignés dans la séquence de la protéine. On peut utiliser encore une fois l’exemple de la ribonucléase. On
a vu que le site actif correspond à 3 acides aminés cruciaux : His12, His119 et Lys 41, sur un total de 124 acides
aminés. Dans cet exemple, l’acide aminé nécessaire à la fixation du substrat est la lysine 41, dont le NH3+ établit
1
2

Métabolisme cellulaire - 5
une liaison ionique avec le groupement phosphate (chargé -) d’un nucléotide. Les deux histidines sont elles à la
base du mécanisme de catalyse (cf. effet du pH).
Le substrat se lie à l’enzyme au niveau du site actif par des liaisons faibles. Un autre exemple caractéristique est la
chymotrypsine. Il s’agit d’une peptidase : elle catalyse l’hydrolyse de la liaison peptidique, mais au niveau d’acides
aminés aromatiques (tyrosine, tryptophane). Le site de fixation de l’enzyme correspond à une poche hydrophobe
(constituée par des acides aminés hydrophobes de l’enzyme) qui interagit avec la partie apolaire du substrat
(interactions hydrophobes). Les acides aminés 193 et 214 établissent des liaisons hydrogène, par leur groupement
peptidique Le site catalytique est constitué de 3 acides aminés Sérine 195, Histidine 57, aspartate 102 (non
représentée). La formation du complexe enzyme substrat repose donc sur des liaisons faibles (toute la panoplie
habituelle) ce qui permet des propriétés de réversibilités, de dynamisme.
De plus, n’oublions pas la stéréospécificité : l’enzyme et le substrat ont des formes complémentaires, ce qui
facilite la mise en place des liaisons faibles. D’où l’analogie très utilisée de la clé et de la serrure. Toutefois cette
analogie néglige un aspect important de la formation du complexe : les modifications induites par le substrat. Par
exemple, l’hexokinase (déjà vue).
Elle catalyse la réaction : Glucose + ATP ADP + glucose 6P
Les études de diffraction aux rayons X ont permis d’étudier la structure de l’enzyme et du complexe enzyme
substrat. La fixation du glucose induit un changement de la conformation spatiale de l’enzyme. Les 2 lobes
entourant le site actif pivotent et enferment le glucose dans une poche. Cette rotation permet par ailleurs
l’exclusion des molécules d’eau du site actif. Il y a deux formes ouverte et fermée. Ce changement a d’ailleurs des
conséquences fonctionnelles importantes : l’ATP n’est pas hydrolysé puisqu’il n’y a pas d’eau dans
l’environnement réactionnel. Le phosphate est bien transféré sur le glucose. Ce qui n’est pas évident a priori
puisque l’hydrolyse de l’ATP est plus favorable (rG=-30,5kJ/mol) que le transfert du phosphate sur un ose (rG=-
13,8kJ/mol).
D’où la dernière caractéristique importante d’un site actif : c’est un site qui le plus souvent exclut les molécules
d’eau. Ainsi le site actif crée des conditions particulières, une sorte de microenvironnement hydrophobe, qui
favorise la réaction (cf. exemple de l’hexokinase)
B. Mécanisme catalytique : exemple de l’oxydation du glycéraldéhyde 3P
La réaction faut partie de la glycolyse qu’on reverra dans la partie suivante.
Glycéraldéhyde3P + Pi + NAD+ 1,3biphosphoglycérate + NADH,H+
Il s’agit d’une phosphorylation, on passe du glycéraldéhyde avec 1 phosphate au biphosphoglycérate (2
phosphates). Mais c’est aussi une réaction d’oxydoréduction : le glycéraldéhyde cède 2e- (et 2 H+) au NAD+.
L’enzyme qui catalyse la réaction est une Glycéraldéhyde3P déshydrogénase. Elle fonctionne avec un coenzyme
qui est le couple NAD+/NADH,H+. (Interaction avec l’enzyme par liaisons faibles)
La réaction globale est exergonique (les potentiels redox sont favorables)
1- Le glycéraldéhyde se lie avec une cystéine de l’enzyme, au niveau du groupement thiol (SH). Le résultat
est un groupement de type hémithioacétal. On remarque qu’on est dans un cas particulier : ici le
complexe enzyme substrat passe par la formation d’une liaison covalente.
2- L’étape suivante correspond à la réduction d’une molécule de NAD+. Cela consiste en un transfert d’ion
hybride H-, c’est-à-dire H avec 2 électrons. Ainsi qu’un H+. L’oxydoréduction correspond bien ici au
transfert de deux électrons. La ½ équation pour le coenzyme est :
NAD+ + H- + H (i.e. 2e-) NADH,H+
Le substrat passe alors à l’état thioester.
3- Le coenzyme réduit est libéré, il est remplacé par un nouveau. Le thioester comprend une liaison « riche
en énergie »
4- La liaison thioester est rompue, ce qui libère de l’énergie (réaction très exergonique). Cette énergie est
utilisée pour la phosphorylation : le 1,3BPG est libéré.
En principe, l’oxydation d’un aldéhyde nécessite le passage d’une barrière importante. On a vu qu’il faut retirer
un ion H-, à l’aldéhyde. Ce qui induirait l’apparition d’une charge + sur le carbonyle. Or, le carbone porte déjà une
charge partielle positive (puisque O est plus électronégatif). La réaction est exergonique (avec NAD) mais il y a
passage par un intermédiaire peu favorable (Ea élevée). La formation du complexe enzyme substrat, par liaison
covalente en l’occurrence, fait apparaître l’hémithioacétal. Lui cède plus facilement l’ion hybride (d’où
l’abaissement de l’énergie d’activation CQFD)
 6
7
8
9
10
11
6
7
8
9
10
11
1
/
11
100%