15/10/2014 TEHHANI Anissa L2 (CR : Hamza Berguigua)

GM – Hérédité mendélienne : Maladies de transmission monogénique dominante, maladies de
transmission monogénique récessive, mosaïques germinales et somatiques.
15/10/2014
TEHHANI Anissa L2 (CR : Hamza Berguigua)
GM
Pr. NGUYEN
14 pages
Les bases fondamentales de l'hérédité
A. Rappels :
L'hérédité est porté par les gènes situés sur les chromosomes (configuration de l'ADN dans les noyaux des
cellules eucaryotes). Le support génétique est composé par des chromosomes au nombre de 46 chez l'Homme,
soit 30 000 paires de gènes ou 3x10 ^9 paires de bases.
Hérédité mendélienne :
Les termes monogénique, mendélienne et monofactoriel sont synonymes : il s'agit d'une mutation dans un seul
gène qui cause la maladie, ces termes sont relatifs car ils sont comparés à l'hérédité polygénique. Le terme
monogénique n'est pas très exact car même dans ces maladies il y a intervention d'autres gènes pour
l'expression, la transmission etc...
Les maladies abordées dans ce cours sont rares et monogéniques contrairement aux maladies plus communes
qui sont aussi plutôt polygéniques.
Il existe aussi des maladies héréditaires à transmission non mendélienne.
Un gène correspond à l'unité d'information génétique.
Un LOCUS désigne la position spécifique d'une séquence d'ADN sur un chromosome et fait référence à la
position d'un gène sur un chromosome. Les gènes autosomiques (chromosomes l à 22) sont présents sur 2
loci homologues, un sur le chromosome d'origine paternelle, un sur celui d'origine maternelle. Tandis que
pour les gènes situés sur le chromosome X, les mâles XY ont un locus (hémizygotes) alors que les femelles
XX ont 2 loci.
Le terme d'ALLELES correspond aux différentes versions d'un gène à un locus particulier. Les allèles
diffèrent entre eux par leur composition nucléotidique ou leur séquence d'ADN (allèle muté VS allèle normal).
On décrit des systèmes bi-alléliques (à 2 allèles différents) et des systèmes multi-alléliques (allant de 3 à 100
allèles différents).
Mutation : variation dans le séquence d'un gène qui est pathologique.
Polymorphisme : variation de séquence par rapport à la normal qui n'a pas de conséquence sur la
fonctionnement du gène.
Allèle morbide : mutation responsable d'une maladie génétique particulière.
1/14
Plan
A. Rappels
B. L'hérédité autosomique dominante
I. Règle théorique de l'hérédité autosomique dominante
II. Exceptions
C. Hérédité autosomique récessive

GM – Hérédité mendélienne : Maladies de transmission monogénique dominante, maladies de
transmission monogénique récessive, mosaïques germinales et somatiques.
On parle d'HOMOZYGOTIE si 2 allèles à un locus sont identiques, on dit que l'individu est homozygote à
ce locus.
On parle d' HETEROZYGOTIE si 2 allèles à un locus sont différents (par exemple l'un est muté).
L'individu est alors hétérozygote à ce locus.
Le GENOTYPE définit la constitution ou la composition génétique d'un individu. On peut
parler du génotype (configuration des allèles) à un locus donné.
Le PHENOTYPE est l'expression du génotype, l' ensemble des caractères apparents permettant de reconnaître
un individu.
Génétique/Congénital :
une maladie congénitale est une maladie présente dès la naissance, une maladie génétique peut être congénitale.
Il existe des maladies génétiques non congénitales (Alzheimer) et d'autres maladies non génétiques mais
congénitales (malformation...).
DOMINANCE : un caractère muté est dominant lorsqu'il entraîne un effet phénotypique même à l'état
hétérozygote.
RECESSIVITE : un caractère est récessif lorsqu'il n’entraîne pas d'effet phénotypique à l'état hétérozygote,
donc uniquement à l'état homozygote. Les notions de dominance et de récessivité ont une influence relative
de 2 allèles l'un par rapport à l'autre au même locus sur des chromosomes homologues.
B. L'hérédité autosomique dominante :
Le gène responsable de la maladie est sur un autosome (donc la maladie ne dépend pas du sexe) et il est
dominant à l'état normal (sauvage chez l'animal). Le phénotype sera anormal à l'état hétérozygote. On utilise ici
un système bi-allélique.
En génétique formelle, on a 3 génotypes possibles pour 2 phénotypes : le phénotype anormal pour le sujet
hétérozygote comme homozygote et un phénotype normal avec aucune mutation sur les 2 allèles. Le caractère
muté dominant a le même effet à l'état hétérozygote qu'à l'état homozygote, en effet il suffit d'une copie mutée
pour que le phénotype s'exprime.
En génétique humaine, c'est-à-dire dans la pratique médicale, le sujet hétérozygote est “simplement” malade
alors que le sujet homozygote portant la mutation sur les 2 allèles a un phénotype beaucoup plus sévère voire
létal, ce dernier cas est rare car il nécessite 2 parents malades et les concernés meurent vite.
2/14

GM – Hérédité mendélienne : Maladies de transmission monogénique dominante, maladies de
transmission monogénique récessive, mosaïques germinales et somatiques.
Le sujet malade possède des gamètes portant l'allèle A et d'autres avec l'allèle a. Il s'unit à un sujet sain de
génotype aa. A la descendance :
•les enfants portant l'allèle A du père associé à l'allèle “a” de la mère sont hétérozygotes pour l'allèle
muté et ont des gamètes sains ou mutés. Avec le génotype Aa, ils sont malades.
•ceux héritant de l'allèle “a” du père sont homozygotes donc tous les gamètes sont normaux, ils ont le
génotype aa et sont sains.
Ainsi, les enfants du père malade Aa ont 1 chance sur 2 d'avoir la maladie et cette probabilité ne dépend pas
du sexe des parents et des enfants.
I. Règle théoriques de l'hérédité autosomique dominante :
On dit que dans une maladie autosomique dominante, la transmission est verticale car la maladie se transmet
de génération en génération.
Un individu atteint a 50% de risque de transmission à sa descendance.
Un individu atteint a forcément un parent atteint.
Un individu sain.
La maladie est indépendante du sexe et du génotype du parent non atteint.
Quelques exemples :
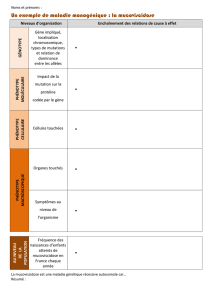
•Maladies osseuses : l'achondroplasie correspond au nanisme le plus commun. Parmi les maladies rares,
elle est relativement fréquente (l/20000). Elle se traduit par un retard de croissance. On observe un
raccourcissement de la racine des membres (bras et cuisses courts, avant-bras et jambes normaux).
C'est une affection congénitale.
•Maladies du tissu conjonctif : la maladie de Marfan (l/5000). Elle se manifeste par une croissance
excessive. Les individus sont longilignes et présentent des problèmes cardiaques, artériels et
ophtalmologiques.
3/14

GM – Hérédité mendélienne : Maladies de transmission monogénique dominante, maladies de
transmission monogénique récessive, mosaïques germinales et somatiques.
•Maladies neurodégénératives : la maladie de Huntington (l/20000) est une démence dégénérative de
début retardé qui commence vers 40 ans. Les individus qui commencent à être malades ont en général
déjà eu des enfants, et donc déjà transmis leur allèle muté. Les descendants qui vont être malades sont
jeunes et sont alors tout à fait sains, bien qu'ils portent la mutation. On peut faire un diagnostic prédictif
pour les descendants.
•Maladies neuromusculaires : la maladie de Steinert (l/8000, DMPK) est une dystrophie musculaire. La
Maladie de Recklinghausen ou neurofibromatose de type 1 (NFl, l/4000) est relativement fréquente
et extrêmement variable. Elle prédispose à développer des tumeurs bénignes de type neurofibromes.
•La Polykystose rénale autosomique dominante (l/l000, PKDl) se traduit par des kystes rénaux.
•Déficits sensoriels : surdités, maladies ophtalmologiques.
•Cancers familiaux (rétinoblastome, polypose colique familiale, cancer héréditaire du sein chez la
femme).
II. Exceptions :
Cas sporadique : comment se fait-il qu'aucun des 2 parents ne soit atteint ?
Saut de génération : 2 cas dans une famille ne peuvent pas relever d'une coïncidence. Comment expliquer que
l'individu « intermédiaire » soit sain ?
Première explication : nouvelle mutation (de novo, néomutation)
Au niveau génotypique, les 2 parents sont indemnes de la maladie et ont un phénotype normal. Ils ne portent
pas de mutations contrairement à leur fille qui a un risque de transmission de 50%. Cependant le risque qu'un
deuxième enfant ait la maladie n'est pas de 50%.
Un accident mutationnel peut arriver à des moments variables au niveau du développement de l' individu.
La mutation survient dans la lignée germinale de l'un des 2 parents.
La mutation apparaît lors de la division cellulaire qui donne les cellules somatiques (saines dans ce cas) et les
cellules germinales (majoritairement normales). Seule une cellule, un clone cellulaire minoritaire est porteur
de la mutation et si cette cellule mutée participe à la fécondation, cela provoque la maladie. L'enfant aura alors
la mutation dans toutes ses cellules d'où le risque de transmission à 50%.
La mutation peut avoir lieu dans la lignée germinale du père ou de la mère, et on ne pourra pas savoir de quels
4/14

GM – Hérédité mendélienne : Maladies de transmission monogénique dominante, maladies de
transmission monogénique récessive, mosaïques germinales et somatiques.
parents il s'agit.
Dans les maladies où les individus atteints ont en général un nombre d'enfants identique à celui de la
population générale, le taux de mutations de novo est très faible. Par exemple la maladie de Huntington
apparaît à l'âge de 40 ans et comme avant les individus ne sont pas malades, ils ont eu le temps de faire un
(des) enfant(s) et la fertilité est identique à celle des personnes non atteintes par cette maladie. On a donc
pratiquement que des cas hérités (l% de mutations de novo).
Dans les maladies dominantes létales comme le nanisme thanatophore, il y a l00% de mutations de novo :
puisque la maladie est létale, les malades ne peuvent pas transmettre leur mutation. Les cas observés sont
forcément issus de mutation de novo.
Le taux est intermédiaire dans les maladies où les individus sont fertiles mais ont moins d'enfants que la
population générale (Achondroplasie = 80 % de mutation des novo donc 8 fois sur 10 les parents sont
normaux), les maladies à expressivité variable (Recklinghausen = 50 %).
En effet les personnes atteintes d'achondroplasie font moins d'enfants donc la fertilité est moindre. Le taux de
mutations de novo est alors plus important. En effet s'il n'y avait pas de néomutations la maladie disparaîtrait
progressivement alors que son taux est constant.
Les mutations de novo (dominantes) sont favorisées par un âge paternel avancé.
5/14
 6
7
8
9
10
11
12
13
14
6
7
8
9
10
11
12
13
14
1
/
14
100%