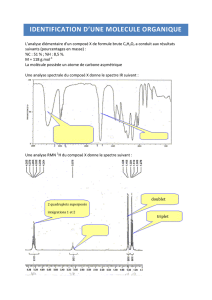
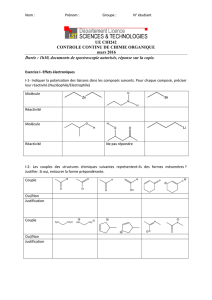
Sérenna Sen Louise Miglianico 25.10.10 Pharmacologie, Anti

Page 1 sur 19
Sérenna Sen
Louise Miglianico
25.10.10
Pharmacologie, Anti-infectieux, les antifongiques, anti – tuberculeux, macrolides et fluoroquinolones, M. O. Tribut
En fin de cours, le prof a dit « si je réactualise mon poly sur le réseau pédagogique, ça voudra dire que j’ai travaillé dessus
donc… que je peux poser des questions dessus, par contre si je ne le réactualise pas, ça veut dire que je n’aurais pas
travaillé dessus donc… que je poserai sans doute pas de questions dessus »
Les formules des molécules ainsi que les doses dans la posologie ne sont pas à connaître.
LES ANTIFONGIQUES
I- Introduction
Les antifongiques (AF) sont des molécules utilisées dans le traitement des lésions dues aux
champignons, c’est-à-dire les mycoses profondes et superficielles, invasives.
La fréquence des mycoses est en constante progression due à la multiplication des facteurs
d’immunodépression (aplasie, greffes, VIH … ). Un AF peut être prescrit en curatif mais aussi en
préventif pour éviter les infections fongiques chez les malades immunodéprimés.
On observe l’émergence d’une grande quantité et variété de champignons opportunistes.
On trouve de plus en plus de souches résistantes aux AF azolés et il existe une toxicité importante
de nombreux AF, c’est pourquoi il est nécessaire de développer de nouvelles molécules.
Dans ce cours, on évoque les AF à action systémique et non locale.
II- Amphotéricine B
A- Structure
L’ampho B est une grosse molécule cyclique avec un noyau macrocyclique lipophile. Cette
molécule est issue de Streptomyces nodosus.
B- Mécanisme d’action
Elle possède un spectre d’action large avec activité fongicide et fongistatique
Elle agit :
- par fixation sur l’ergostérol membranaire et par augmentation de la perméabilité
membranaire
- par la formation de pores
- par la stimulation de la consommation d’O2, dégradation d’ATP, diminution des
synthèses et fuite des métabolites essentiels

Page 2 sur 19
C- Pharmacocinétique
L’ampho B possède une absorption digestive nulle expliquant une administration par IV. Elle
circule grâce à une forte liaison protéique (95%) et peut se stocker dans les tissus entrainant un
relargage secondaire dans la circulation générale.
Elle diffuse faiblement dans le LCR.
Sa demi-vie est longue (24-48h).
Son métabolisme est mal connu mais on sait qu’elle est faiblement éliminée par voie rénale.
D- Effets indésirables
L’ampho B peut induire :
- fréquemment une insuffisance rénale avec réduction de la filtration glomérulaire
- hypokaliémie par fuite urinaire
- toxicité hématologique (anémie, thrombopénie, granulopénie)
- effets neurologiques (nausées, céphalées, vertiges)
- fièvre fréquente
E- Contre-indications
Il existe peu de risque avec cette molécule.
On peut cependant trouver des contre-indications dans des cas : d’allergie, d’insuffisance rénale,
grossesse ou allaitement.
F- Résistance et associations
Il existe un risque négligeable de contracter des mutants résistants au cours des traitements
prolongés car il existe une synergie habituelle avec la 5-Fluorocytosine.
G- Formes galéniques
FUNGIZONE ®
L’administration de la molécule se trouve sous deux formes :
- forme injectable : pour les infections fongiques systémiques
- forme per os : pour les atteintes oro-pharyngées
L’ampho B a la caractéristique de rester dans la circulation et de diffuser dans les graisses, elle
arrive ensuite au rein où elle provoque une toxicité. C’est pourquoi depuis quelques années, on
associe cette molécule à des complexes lipidiques (liposomes ABELCET® et AMBISOME®) qui
permettent d’augmenter la solubilisation donc de diminuer les effets secondaires dont la toxicité
rénale. Une administration de dose plus importante sera donc possible.

Page 3 sur 19
III- Fluorocytosine
A- Structure
La 5-fluorocytosine (5FC) est une petite molécule, analogue des bases nucléotides : elle
correspond à un noyau pyrimidine (cytosine) fluoriné en 5.
B- Mécanismes d’action
Elle possède un spectre étroit avec une action sur Candida, Cryptococcus neoformans et certains
agents de chromomycose.
La 5FC passe dans les cellules grâce à une cytosine perméase et est transformée par une cytosine
désaminase en 5-fluoro uracile (5FU) toxique. Celui-ci perturbe la synthèse des protéines par
substitution à l’uracile dans l’ARN fongique, le 5FU est un inhibiteur compétitif face à l’uracile
fongique.
De plus, la 5FC empêche la synthèse de l’ADN fongique par inhibition de la thymidilate
synthétase.
C- Pharmacocinétique
Cette molécule est administrée :
- par voie PO : rencontre avec des bactéries digestives pouvant transformer la 5FC en
5FU donc augmentant la toxicité
- par voie IV : pas d’interaction avec les bactéries digestives, la pro-drogue est donc
préservée amoindrissant la toxicité
Les concentrations sériques optimales sont atteintes en 30 min à 2h.
Cette molécule possède une bonne diffusion dans l’organisme notamment dans le LCR
(concentration LCR concentration sérique) impliquant un risque de neurotoxicité.
Elle ne se lie pas aux protéines et possède une élimination urinaire (la posologie doit être adaptée
en prenant en compte la fonction rénale, il existe une bonne corrélation entre la clairance de la
créatinine et la 5FC).
D- Effets indésirables
Il existe des effets secondaires mineurs tels que la nausée, vomissement, diarrhée.
D’autres plus graves : troubles hématologiques, hépatiques.
Ces effets sont observés en cas de prise prolongée.
E- Contre-indications
On trouve des contre-indications chez les sujets en aplasie (toxicité hémato), femmes enceintes et
une adaptation est nécessaire pour les patients en insuffisance rénale.
Ces contre-indications sont à pondérer, en effet, il est indispensable d’évaluer le rapport bénéfices-
risques attendus (que faire pour un sujet aplasique risquant une infection opportuniste ?).

Page 4 sur 19
F- Résistance et associations
Des souches résistantes peuvent apparaître notamment dans les Candida albicans de sérotype B et
chez les Candida tropicalis.
Il faut éviter de le prescrire en monothérapie et plutôt l’associer avec une molécule comme
l’ampho B ou un dérivé azolé.
G- Formes galéniques
ANCOTIL® en comprimés ou flacon injectable
Cette molécule est prescrite contre des mycoses systémiques ou des phanères.
IV- Dérivés azolés
A- Structure générale
Ce sont des molécules de synthèse issues de la modification du noyau azolé.
Les premiers utilisés : les imidazolés, forts inhibiteurs enzymatiques entrainant de nombreuses
interactions, le plus souvent utilisé PO.
Plus récemment : les triazolés (fluconazole, itraconazole…)
B- Mécanisme d’action général
Il agit au niveau de la membrane des champignons en bloquant le cytochrome P450 des
mitochondries servant à la synthèse de l’ergostérol membranaire ce qui aboutit à l’inhibition de la
construction de la membrane des champignons.
C- Fluconazole
1- Structure
Cette molécule est composée d’un noyau triazolé.
Elle a la caractéristique d’être plus hydrosoluble et peu lipophile par rapport aux autres azolés, lui
conférant une meilleure diffusion et une meilleure absorption (moins de variabilité de l’effet).
2- Spectre
Le fluconazole possède une excellente activité sur Candida albicans et est actif contre certains
Candida non albicans, il est donc indiqué principalement lors de candidose.
Il est aussi actif sur Cryptococcus neoformans.
On peut trouver quelques souches de Candida résistantes.

Page 5 sur 19
3- Pharmacocinétique
Il peut être administré PO ou par IV.
Il diffuse facilement dans les tissus et se lie faiblement aux protéines plasmatiques.
Sa demi-vie est de 24h.
Son élimination est urinaire, une adaptation posologique chez les insuffisants rénaux est conseillé
car il présente une plutôt bonne tolérance.
4- Effets indésirables
Cette molécule est en général bien tolérée mais on peut assister à des effets indésirables mineurs
(troubles gastro-intestinaux, rares céphalées et rares éruptions cutanées).
5- Interactions médicamenteuses et contre-
indications
Par son inhibition du cytochrome P450, le fluconazole peut modifier la pharmacocinétique de
certains médicaments (immunosuppresseurs…) :
- élévation des concentrations sériques par inhibition du métabolisme de la phénytoïne
(anti-épileptiques et anti-arythmiques cardiaques)
- potentialisation des agents hypoglycémiants oraux et la warfarine
- diminution des concentrations sériques et de l’efficacité des deux molécules lors de la
prescription conjointe avec de la rifampicine
- augmentation des effets immunosuppresseurs et de la créatininémie lors de la prescription
avec de la ciclosporine et le tacrolimus
Il est contre-indiqué chez les femmes enceintes.
D- Itraconazole
1- Spectre
L’itraconazole a un spectre large, il est actif sur : les levures, les dermtophytes et sur de
nombreuses moisissures, notamment l’Aspergillus.
2- Pharmacocinétique
Il existe une différence entre l’administration PO et par IV (car pas de corrélation entre les doses),
ce qui complique le passage d’un traitement par IV (de première intention) à un traitement PO.
On trouve une grande variabilité inter-individuelle.
Cette molécule est fortement liée aux protéines plasmatiques.
Elle est lipophile, donc diffuse dans les tissus graisseux et s’y stocke devenant ainsi inactive : elle
est moins ubiquitaire que le fluconazole.
Sa demi-vie est assez longue : 20-30h.
Son métabolisme est essentiellement hépatique et un de ses métabolites (l’hydroxy-itraconazole)
est actif.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
6
7
8
9
10
11
12
13
14
15
16
17
18
19
1
/
19
100%