Analyse des gènes du MisMatch Repair dans le syndrome de Lynch

104Cancéro dig. Vol. 2N°2-2010- 104-110
10.4267/2042/32416
© aln.editions
MISE AU POINT
Analyse desgènesduMisMatchRepairdansle syndrome de Lynch
MisMatchRepairgenesand Lynchsyndrome,recent updates
Sylviane Olschwang, FrançoisEisinger
Institut Paoli-CalmettesetCentre de Recherche en Cancérologie de Marseille (INSERM UMR891),
232, boulevardSainte-Marguerite, F-13009Marseille
sylviane.olschwang@inserm.fr
❚Résumé
Lesyndrome de Lynch est dûàune mutation constitutionnelle
d’un gène MMR (MisMatchRepair),etest la cause de 0,9à3,4 %
descancers colorectaux.Outre le faitquecescancers se
développentplus précocementque danslapopulation générale,
lesmutationsaugmententaussi le risque de cancers gynéco-
logiques(endomètre,ovaire),des voies urinaires,etde l’appareil
digestif (intestin grêle,estomac, voiesbiliaires). Lerecueil des
éléments cliniques,généalogiquesetbiologiques,est
indispensable pour identifierlespersonnesporteusesd’un
syndrome de Lynch,lesrecommandations spécifiquesde prise
en charge ayantfaitlapreuve d’une réduction quasi-complète de
lamortalité danscette pathologie. Enfin,danslamesure oùles
pratiquesmédicalesdiffèrentparfoisde manière importante entre
Centres,en termesd’accessibilité,d’acceptabilité etde règles
d’usage,d’importants efforts d’harmonisation sontfaits àl’échelle
nationale eteuropéenne.
Mots-clés
Syndrome de Lynch, GènesMMR, CancerMSI, Mutation constitutionnelle, Examen génétique
❚Abstract
Lynchsyndrome iscaused byinherited germline mutationsin MMR
(MisMatchRepair) genes,thataccountfor 0.9-3.4% of colorectal
cancers.Inaddition toyoung onsetcolorectalcancer,itischaracte-
rized byan increased risk of gynaecologic(endometrium,ovary),
urinary tractand extracolonicgastrointestinalcancers (small intestine,
biliary tractand stomach). Tumor,patientand familyrecordsare
crucialto earlyidentifyindividualsat risk forLynchsyndrome. Genetic
test results can guide screening recommendationsforpatients and
theirfamilies, as specific clinical guidelineshavebeen proved to
reduce morbidity and mortality in thisdisease. Asheterogeneous
attitudesin clinical practice of genetictesting exist among countries
in termsof access, acceptance,useand rules,harmonizing clinical
genetictesting servicesprogressivelyimplementrelevantindication
criteriaforgenetictesting.
Keywords
Lynch (HNPCC)syndrome, MMR genes, MSI tumor type, Germline mutation, Genetictesting
❚Introduction
Lespersonnesporteusesd’une mutation constitutionnelle d’un
gène MMR (DNA MisMatchRepair system) constituent un groupe
de lapopulation dontl’identification permet une prise en charge
efficace du risque[1].Cespersonnesontdes risquesrelatifset
absolus trèsélevésde développer uncanceren l’absence de
prévention spécifique. En effet,une mutation constitutionnelle d’un
gène MMR, c’est-à-dire la constitution génétique d’une personne,
est un facteur de risque importantde cancer ; lesconséquences
phénotypiques, c’est-à-dire lescancers eux-mêmes,en sontla
traduction clinique,l’ensemble de cesmanifestationsétantnommé

Cancéro dig. Vol. 2N°2-2010105
GènesMMR (MisMatchRepair):groupe de gènesapparentés
pour leur fonction de contrôle de l’intégrité de l’ADN lors de sa
réplication en mitose. Lesmutationsde quatre membresde cette
famille,MLH1,MSH2,MSH6etPMS2,sontresponsablesd’un
syndrome de Lynch. D’autresmembresontété étudiés(PMS1,
MLH3and EXO1) maisnon retenus comme directementimpliqués
dansle syndrome de Lynch.
❚Lesoutils
L’absence de phénotype spécifiqueau syndrome de Lyncha
conduità analyser un ensemble de paramètresqui,seulsouen
combinaison,sont utilesàl’évaluation diagnostique.
Lesinformationsindividuelles
Les risquesrelatifsetabsolus de cancers,en casde mutation
constitutionnelle d’un gène MMR, sontmaintenantbien connus
et varientpeud’une étude àl’autre[8,9].La probabilité de trouver
une mutation constitutionnelle d’un gène MMR a égalementété
trèsétudiée pour validerlesindicationsd’analyse génétique[9].
Cesindications sesontprogressivementélargiespour intégrerles
patients ayant une atteinte,soit sporadiqueavant40ans,soit
s’accompagnantd’unantécédentpersonnel oud’unantécédent
aupremierdegré de cancer,toutesces situationsentraînant une
probabilité de trouver une mutation constitutionnelle d’un gène
MMR supérieureà20 %. La localisation descancers,outre les
4 principales, a été étendueàl’ovaire, aux voiesbiliairesetà
l’estomac dontles risquesrelatifsdépassent5.
Informationsfamiliales
L’histoire familiale est un pointcentral de l’évaluation du risque de
prédisposition génétique. La probabilité de trouver une mutation
augmenteavecle nombre de parents atteints d’uncancerdu
spectre de Lynch, ce d’autantque lalocalisation tumorale
concerne un organe pour lequel le risquerelatif est élevé. Par
exemple,lameilleurevaleur prédictive est donnée parl’adéno-
carcinome de l’intestin grêle dontle risquerelatif dépasse25,puis
les voies urinaires(RR >20) etle côlon/rectum (RR > 10)[10].
Cependant,lespersonnesporteusesde mutation serontplus
souventrepéréespar une agrégation familiale de cancers
colorectaux,situation laplus fréquente. Elle augmente également
lorsque le cancerapparaîtàunâge significativementplus jeune
quecelui observé danslapopulation générale,lorsque le nombre
de parents non atteints est faible auregard de leur âge, avecune
proximité généalogique,etlorsque le cancer semble obéiràune
ségrégation autosomique dominante.
Informations tumorales
❙Génotypage de l’ADN microsatellite
àlarecherche d’une instabilitéMSI
Lescancers survenantchezlespersonnesporteusesd’un
syndrome de Lynch ont une caractéristiqueacquiseàlasuite de
«Syndrome de Lynch ». Cesyndrome est repérable par une
anomalie génétique descellulescancéreuses,l’instabilité de l’ADN
microsatellite, appelée « MSI » qui est une conséquencesystéma-
tique de l’absence de fonction MMR.
En pratique médicale,lasituation est plus complexe. Leterme
de «Syndrome HNPCC » (Hereditary Non-PolyposisColon/
ColorectalCancer)caractérisaitinitialementl’existencechez un
patientde critèrescliniques, connus sous le nom de « Critères
d’Amsterdam »,proposésen 1991 etrevus en 1999, auxquels
ontété intégrésdescritèresmoléculaires, comme l’anomalie MSI
parexemple,en 1998. Cesyndrome est apparu progressivement
comme une entité hétérogène,etle terme même est délaissé.
Parmi lespatients ayantlescritèrescliniquesdu syndrome
HNPCC, environ untiers ont une fonction MMR intacte,sansMSI
danslescellulescancéreusesni mutation constitutionnelle d’un
gène MMR.Cettesituation aété individualisée récemment,dans
le syndrome HNPCC, sous le terme de « CancerColorectal
FamilialType-X»[2,3]dûàdesmutationsde gènesinconnus,ou
àune combinaison de caractéristiquesgénétiquesetenvironne-
mentalesdontl’évaluation n’est pasaccessible actuellement,
ouencoreàun défaut de sensibilité des techniquesanalytiques.
Cesyndrome « X» n’est pasl’objetde laprésentesynthèse. La
situation laplus fréquenteconcerne lespatients chezquiune
mutation constitutionnelle d’un gène MMR est identifiée,souvent
audécours de lamise en évidence d’une anomalie MSI dansles
cellulescancéreuses,etqui donc, même si l’histoire personnelle
etfamiliale ne remplitpasl’ensemble descritèrescliniques,sont
porteusesdu syndrome de Lynch[4].
Plusieurs étudesen population ontpermisd’établirla contribution
desmutationsconstitutionnellesdesgènesMMR à 0,9à3,4 %
descancers colorectaux [5], une proportion qui ne justifie pasde
proposer une recherche systématiqueaux patients porteurs d’un
cancercolorectal. Des stratégiesd’identification ontété progres-
sivementdéveloppéesetévaluées[6,7].La synthèse proposée ici
metl’accent sur lesarguments stratégiquesdansladémarche
d’identification d’une mutation constitutionnelle d’un gène MMR,
àtravers laprésentation desdifférents outilsdisponibles,et
l’intérêtde leur combinaison pour perfectionnerlaméthode
globale d’analyse. Elle comporte,en préambule,une synthèse de
lapartie introductive et,en guise d’information,lastructure du
réseaud’identification du syndrome de Lynch en France.
❚Définitionsdes termesessentiels
Syndrome HNPCC (Hereditary Non-PolyposisColon/Colorectal
Cancer):description clinique etépidémiologique de familles selon
descritèresde nombre,d’âge etde parenté despatients porteurs
de cancers colorectaux (critèresd’Amsterdam définisen 1991 et
revus en 1999).
Syndrome de Lynch: conséquencescliniquesde laségrégation
familiale d’une mutation constitutionnelle d’un gène MMR.
Cancercolorectal familialType-X:syndrome HNPCC non associé
àune mutation constitutionnelle d’un gène MMR.

106 Cancéro dig. Vol. 2N°2-2010
❚Analysescombinées
L’ensemble de cesinformationsest intégré danslesprogrammes
prédictifs,etleur obtention faitl’objetd’arbresde décision
progressifs.
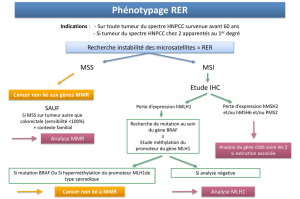
Analyse de lafonction MMR descellules tumorales
Plusieurs stratégiesd’analyse descellules tumoralesontété
proposéesen vue de l’identification d’une mutation constitution-
nelle d’un gène MMR, etdoncd’unsyndrome de Lynch. Elles
diffèrentessentiellement sur l’ordre desdifférentesétapes,mais
latrèsbonne sensibilité dugénotypage aprogressivementfait
préférerla chronologie suivante:
1) Génotypage de l’ADN à larecherche d’une anomalie MSI.
2)Étude immunohistochimique de l’expression des4 protéines
MSH2, MLH1, MSH6etPMS2en casde MSI.
3)Analyse ducodon 600 dugène BRAF en casd’absence de la
protéine MLH1.
La sensibilité etlaspécificité de cettestratégie pourraientêtre
encoreamélioréesen couplantladernière étape àune quanti-
fication de laméthylation dupromoteur dugène MLH1[21];
néanmoins, cettetechnique délicate est difficilementapplicable
en dépistage.
Cetteanalyse doitêtreréalisée en rapport avecle contexte
individuel etfamilial, autantque possible avant,ou, à défaut, au
cours de l’analyseconstitutionnelle (voirplus bas). L’intérêtdu
couplage aété largementdiscutécesdernièresannées[22-24].
Certainesméta-analysesonten effet rapporté l’existence de
mutationsconstitutionnellesassociéesàdes tumeurs MSS ou
danslesquelleslesdeux protéinesMSH2etMLH1 étaient
présentes.Cependant, cesmutationsontététoutesreclassées
en polymorphismesou variationsde signification fonctionnelle
inconnue,grâceàune revuesystématique parle comité interna-
tional de lasociétéINSiGHT qui enregistre l’ensemble desinfor-
mationsen lien avecla base de donnéesinternationale de
mutations, accessible àl’adresse:http://chromium.liacs.nl/
LOVD2/colon_cancer/home.php?action=switch_db.Lesétudes
lesplus récentesrecommandentde réaliserl’analyseconstitu-
tionnelle uniquementaprèsévaluation de lafonction MMR des
cellules tumorales[25,26].De plus,il est recommandé en France
quecette évaluation tumorale soit systématique dèsqu’uncancer
associé au syndrome de Lynch est diagnostiquéavantl’âge de
60 ans,ouen présence d’untel antécédent tumoralchez un
parentaupremierdegré;l’information est,de ceseul fait,dispo-
nible avantque l’indication d’une analyseconstitutionnelle
soitdiscutée (http://www.e-cancer.fr/soins/prises-en-charge-
specifiques/oncogenetique).
Contribution respective desgènesMMR
Lesdéfaillancesdu système MMR, misesen évidence parle
développementde cancers MSI, sontessentiellement(comme vu
plus haut) duesàune méthylation acquise dupromoteur dugène
l’inactivation d’un gène MMR, l’instabilité de l’ADN microsatellite,
ouMSI ;cettecaractéristique est détectée par un examen
génétiquesimple aumoyen d’un kitcommercial qui permet
l’analyse de l’ADN microsatelliteauniveaude 5 loci « poly-A»
retenus dès1998 parconsensus [11].Cetexamen appelé
« génotypage » est réaliséaussibien sur tissu tumoral préala-
blementfixéàpHneutre oucongelé aprèsintervention chirur-
gicale oubiopsie. Il est indispensable,pour garantirlafiabilité de
l’examen,de sélectionner une zone riche en cellules tumorales,
sous contrôle microscopique d’une coupe histologiquecolorée.
❙Méthylation dupromoteur dugène MLH1
Environ 10% descancers colorectaux sporadiquesont untype
MSI, maismoinsd’untiers d’entre eux traduit unsyndrome de
Lynch. L’identification d’une tumeur MSI ne justifie doncpas,à
elle seule (c’est-à-dire en l’absence de prise en compte de la
présentation clinique),de proposer une analyseconstitutionnelle
desgènesMMR.La majorité descancers MSI est la consé-
quence d’une absence d’expression dugène MLH1parméthy-
lation de son promoteur,fréquente danslescellulesépithéliales
coliques sénescentes.Cependant, ce mécanisme est également
observé en présence d’unsyndrome de Lynch,dansenviron un
tiers descancers,etn’est doncpas suffisamment spécifique
pour être proposécomme test discriminant,même si des varia-
tions subtilesdes sitesde méthylation différencientlesdeux
situations[12].
❙Mutationsdugène BRAF
La caractérisation dugène BRAF permetd’améliorerlasélection
des tumeurs MSI en rapport avecunsyndrome de Lynch.
L’activation de l’oncogène BRAF parmutation est trouvée dans
de nombreux cancers et,en particulier,15 % descancers
colorectaux [13].Une mutation,p.Val600Glu,est trèsmajoritai-
rement trouvée dans 30 % descancers MSI et10% descancers
MSS [14].Dansles tumeurs MSI, elle concerne quasimentexclu-
sivementcellesconsécutivesàune méthylation dupromoteur
dugène MLH1[15] aucontraire de cellesen relation avecun
syndrome de Lynch[16,17].L’incorporation de larecherche de
cette mutation dansl’algorithme d’investigation du syndrome de
Lynchadoncétérecommandée [18].Cetteanalysetrès simple
permeten effet une réduction de 20 % desdemandesd’analyse
desgènesMMR dontl’efficacitéatteint 60-70 % lorsqu’une
sélection préalable des tumeurs avec absence d’expression du
gène MLH1 est appliquée [19].
❙Immunohistochimie
L’utilisation d’anticorpscommerciaux reconnaissantlesquatre
protéinesMMR est nécessaire pour identifierla/lesprotéine(s) non
fonctionnelle(s),etle degré de déficitqui peut,danscertaines
situations,être partiellementcompensé[20].L’inactivation de la
protéine MSH2ouMLH1 entraîne fréquemmentl’absence des
deux protéinesde l’hétérodimèrecorrespondant, à savoirMSH2/
MSH6ouMLH1/PMS2, à l’inverse de laprotéine MSH6ou
PMS2,dontl’absence peut être isolée (Fig. 1).

Cancéro dig. Vol. 2N°2-2010107
MSH6mutationsponctuelles5%
PMS2mutationsponctuelles 2 %
MLH1 mutationsponctuelles
30%
MSH2mutationsponctuelles
30 %
MSH2 réarrangements
génomiques13%
EPCAM 3' délétion 4 %
Autres(MSH6ouPMS2
réarrangements génomiques…)
11 %
MLH1 méthylation 1 %
MLH1réarrangements
génomiques4 %
MLH1 .Dansle syndrome de Lynch,l’origine de cette défaillance
est consécutiveàlaprésence d’une altération constitutionnelle de
l’un desquatre gènesdontl’implication respective n’est pas
équivalente (Fig. 2). Desmutationsponctuelleshétérozygotes sont
trouvéesen égale proportion sur lesdeux gènesMSH2etMLH1 ,
àsavoir 30-35 % chacun. Desdélétionsouduplications
génomiques sont trouvéesdans16-17% descas,4 foisplus
fréquemment sur MSH2quesur MLH1.Lesmutations sur les
gènesMSH6etPMS2sontbeaucoup moinsfréquentes,respec-
tivement 2-10et 2 %,etil aétéconvenude manièreconsensuelle,
pour cesdeux gènes,queseuleslesmutationsinterrompantla
synthèse protéique devaientêtreretenuescomme responsables
du syndrome de Lynch. Enfin,de raresméthylationsdupromoteur
dugène MLH1ontété décriteschezdespatients atteints de
manièresporadiqueàunâge trèsjeune [27,28].Aucune trans-
mission héréditaire n’est,en revanche,documentée [29].Très
récemment,desdélétionsde larégion C-terminale dugène
EPCAM/TACSTD1 ,localisé danslarégion 5’ dugène MSH2 ,ont
été misesen évidenceavec, pour conséquence,laméthylation du
promoteur dugène MSH2et son absence d’expression, cette
Figure2
Contribution respective desgènesMMR au syndrome de Lynch. L’implication des
différentesaltérationsconstitutionnellesdesgènesMMR a été miseàjour en
2009 parSheng etal.,dans une analysesynthétique de l’ensemble desétudes
publiées[46].
Figure 1
Représentation simplifiée du système de réparation desmésappariements de l’ADN.L’ADN normal est formé de 2brinscomplémentairesenroulésen hélicesdontla
séquence,composée des4 nucléotidesA, T, GetC, est plus oumoinscomplexe. L’ADN microsatellite est une répétition de séquencescourtesetpeucomplexes,par
exemple poly-CA/GT (aumilieu) oupolyA/T(àdroite). Aucours de laréplication,l’ADN polymérase intègre lesbasescomplémentairesaubrin parental déroulé pour
synthétiserle second brin. À cette étape,une erreur d’intégration peut fréquemment survenir,particulièrementdansles séquences répétéespeucomplexes,ce qui
provoqueun mésappariement sansbloquerlasynthèse. Le mésappariementest reconnuparlaprotéine MSH2quis’yfixe et recruteson partenaireMSH6ouMSH3
selon le type de mésappariement,maisde manière non exclusive. L’étape de réparation proprementdite faitintervenirMLH1avecd’autresprotéineshomologuesPMS1,
PMS2ouMLH3.En présence de l’hétérotétramère,lesendo- etexonucléasesextraientlaséquence d’ADN incorrecte,etl’ADN polyméraseréintègre de nouveaux
nucléotides.

108Cancéro dig. Vol. 2N°2-2010
niveau tumoral est tout àfaitdiscutable,quellesquesoientles
caractéristiquesde l’histoiretumorale personnelle etfamiliale.
De plus,en présence de donnéesimmunohistochimiques,les
modalitésd’analyseconstitutionnelle deviennent:
–étude première dugène MLH1en casde perte d’expression
isolée de laprotéine MLH1oude perte d’expression des
protéinesMLH1etPMS2auniveau tumoral;
–étude première dugène MSH2en casde perte d’expression
isolée de laprotéine MSH2oude perte d’expression conjointe
desprotéinesMSH2etMSH6auniveau tumoral;
–étude première dugène MSH6en casde perte d’expression
isolée de laprotéine MSH6auniveau tumoral;
–étude première dugène PMS2en casde perte d’expression
isolée de laprotéine PMS2auniveau tumoral.
❚Organisation du réseaunational
en France
Les stratégies sontgénéralementmisesen place en fonction des
contrainteslocaleset,pour le mieux,organiséesàl’échelle
nationale. Ellesimpliquent,dans tous lescas,une expertise
clinique,génétique (médecinsgénéticienset/ouconseillers
génétiquesdiplômés) etplus largementmédicale qui permet une
connaissance parfaite de lapathologie etde lagestion des
risquesassociés,une expertise histologique etmoléculaire[41],
pour laréalisation etl’interprétation desexamens,et une expertise
technique pour laréalisation desexamensde dépistage
(chromoendoscopie parexemple). Lescontrainteslégales
diffèrent significativemententre lespays [42,43], ce d’autantque
lamajorité desanalysesaété initialementpratiquée dansdes
structuresde recherche [44].Enfin,descritèresd’ordre psycho-
logique etl’accessibilitéaux centresaccréditésdoiventêtre pris
en considération [45].
EnFrance,dèsl’identification desdeux gènesprincipaux,MSH2
en 1993etMLH1en 1994,quelqueslaboratoiresd’oncogéné-
tique moléculaire ontproposé d’analysercesgènesavecl’objectif
d’intégrercetteanalyse danslaprise en charge médicale des
patients concernés.Ilsétaientaunombre de 3en 1995,6en
1998,et unréseaupermettantde discuteretd’harmoniserles
pratiquesaétécréé en 2000.
Deux ansplus tard,le ministère de la Santé prenaitladécision de
financerlesactivitésd’oncogénétique,relayé parl’Institut National
duCancer(INCa). Entre2002 et 2009,une nouvelle facette de
l’oncogénétiquea chaqueannée été financée :25 laboratoiresen
2002,l’élaboration de recommandationsnationalesd’identifi-
cation etde prise en charge du syndrome de Lynch[9]etde la
prédisposition aucancerdu sein etde l’ovaire (BRCA) en 2003,
48 structuresde consultation rayonnant sur 102 sitesdans
66 villesen 2004. Leslaboratoiresont vu leur activitérenforcée en
2005,puisétendue grâceàla création de réseaux de prédisposi-
tions raresaudéveloppementde tumeurs en 2005,etlamise en
place de basesnationalesdesmutationsdesgènesMMR et
BRCA en 2006.Le développementde laformation desconseillers
altération étant transmissible [30-32].Cesanomaliesde méthy-
lation despromoteurs de MLH1etMSH2semblentcontribuer,de
manière non négligeable, au syndrome de Lynch[33].
Modèlesprédictifsd’une altération constitutionnelle
d’un gène MMR
À côté de larévision desrecommandationsde Bethesda, cinq
modèlesontété développéspour évaluerl’existence d’un
syndrome de Lynchchez un patient, connus sous les termesde
Leiden [34], MMRpredict[35], PREMM (1,2)[36,37]etMMRpro
[38].À ce jour,troisétudesontcherché à comparerl’efficacité des
différents modèles sur des sériesimportantesde patients atteints
de cancercolorectal,etn’ontpasmisen évidence de différence
importante, avecune performance légèrement supérieure pour
MMRpro[25,39,40].Cependant,l’étatde lafonction MMR dans
lescellulescancéreusesn’est généralementpasconsidéré dans
cesmodèlesdontl’intérêtest essentiellementlimitéaux situations
danslesquellesle génotypage de l’ADN tumoral n’est pas
possible.
Analyseconstitutionnelle
L’indication d’analyseconstitutionnelle desgènesMMR est
classiquementretenue lorsque laprobabilité de trouverapriori
une altération est supérieureà20 %. Àl’inverse,lorsquecette
probabilité est inférieureà10%, aucune analyseconstitutionnelle
n’est proposée. Les situationsintermédiairesimposentl’obtention
d’informationscomplémentaires,tellesque l’étatde lafonction
MMR danslescellules tumorales si elle n’est pasdéjàdisponible.
Enrésumé,parordre de fréquence décroissante,lesmutations
ponctuellesdesgènesMSH2etMLH1arriventen tête,suivies
des réarrangements génomiquesdesgènesMSH2,MLH1et
EPCAM,puisdesmutationsponctuellesdesgènesMSH6et
PMS2et,enfin,de laméthylation dupromoteur dugène MLH1 .
Sur cettebase,laséquence d’investigation desgènesMMR
suivante est proposée :
1) Recherche de mutation ponctuelle sur les régionscodantes
desgènesMSH2etMLH1par séquençage oupré-criblage.
2)Recherche de réarrangementgénomique desgènesMSH2,
MLH1etEPCAM en une étape unique parMLPA avecun kit
commercial.
3)Encasde négativité de cesdeux premièresétapes,il est
recommandé de calculerlaprobabilitérésiduelle,aposteriori,
d’implication desgènesMMR avantde programmerl’analyse du
gène MSH6ouPMS2.
Cependant, aveclarecommandation de mise en œuvre d’une
étude tumorale dans toutesles situationscliniquesévocatrices,et
à chaque foisqu’elle est possible (matériel tumoral disponible et
exploitable), cette proposition est adaptée aux seules situations
danslesquellesl’étude tumorale s’est limitée augénotypage ou
n’apasété possible ;en effet, comme nous l’avonsindiqué
précédemment,lamise en place de l’analyseconstitutionnelle
desgènesMMR en l’absence d’instabilité desmicrosatellitesau
 6
7
6
7
1
/
7
100%