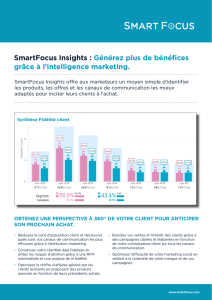
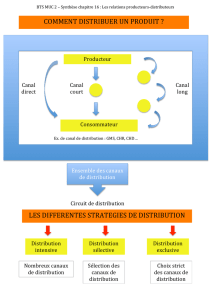
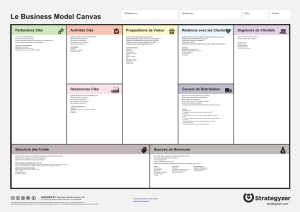
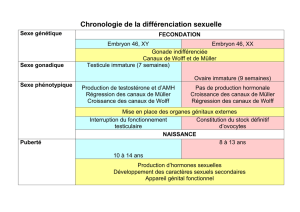
Lire l`article complet

110
La Lettre du Pharmacologue - Volume 19 - n° 4 - octobre-novembre-décembre 2005
DOSSIER
Les canaux calciques voltage-dépendants
comme cibles d’analgésiques
Voltage-dependent calcium channels as targets for analgesics
●
J. Nargeot*, E. Bourinet*
RÉSUMÉ.
Les ions calcium sont impliqués dans de nombreuses fonctions cellulaires allant du couplage excitation-contraction musculaire à la
libération d’hormones ou de neurotransmetteurs et à l’expression de divers gènes. L’entrée rapide de calcium dans les cellules excitables est
régulée principalement par des molécules de la membrane plasmique ou “canaux calciques potentiel-dépendants” qui constituent une classe
hétérogène de protéines sensibles aux variations du potentiel membranaire. Les études fonctionnelles et structurales ont révélé la très grande
diversité de ces canaux, permettant de mieux comprendre comment les cellules utilisent “la transitoire calcique” pour contrôler des fonctions
physiologiques aussi variées. Les canaux calciques neuronaux, qui permettent en particulier la libération des neurotransmetteurs dans le
système nerveux et qui contrôlent l’efficacité synaptique, sont largement exprimés dans les structures impliquées dans la transmission et la
mémorisation de la douleur. Les douleurs neuropathiques se caractérisent par une hyperexcitabilité des neurones nociceptifs accompagnée
d’une baisse des seuils nociceptifs (hyperalgie) et/ou d’une réponse douloureuse à des stimuli normalement non nociceptifs (allodynie). Des
molécules se liant à la sous-unité
α
2-
δ
des canaux calciques (
α
2-ligands) sont largement utilisées dans le traitement des douleurs neuropa-
thiques. Leur mécanisme d’action est probablement lié à une diminution de l’activité des canaux calciques neuronaux et, en conséquence, à
une réduction de l’hyperexcitabilité synaptique. Des études récentes démontrent aussi que les canaux calciques N et T sont impliqués dans la
transmission des messages douloureux et représentent des cibles pharmacologiques d’intérêt thérapeutique.
Mots-clés :
Canaux calciques voltage-dépendants - Douleurs neuropathiques -
α
2-ligands.
ABSTRACT.
Calcium ions mediate a wide range of cellular functions including skeletal and cardiac muscle excitation-contraction coupling,
neurotransmission, activation of calcium-dependent enzymes and gene transcription. The rapid influx of calcium ions into excitable cells is
mainly regulated by molecules of the plasma membrane named “voltage-dependent calcium channels” which constitute an heterogeneous class
of proteins sensitive to variations of the membrane potential. Functional and structural studies have revealed the large diversity of the calcium
channel family allowing a better understanding of how cells can generate appropriate calcium transients to control various physiological
functions. Neuronal calcium channels underly the release of neurotransmitters, control synaptic efficiency in the nervous system and are
largely expressed in the structures involved in the transmission and memorization of pain signals. Neuropathic pain is characterized by an
hyperexcitability of nociceptive neurons accompanied by a decrease of nociceptive thresholds (hyperalgia) and/or a painful response to
normally non-nociceptive stimuli (allodynia). Some molecules binding to the
α
2-
δ
subunit of the calcium channel complex (
α
2-ligands) are
widely used in the treatment of neuropathic pain. Their mechanism of action is probably linked to a decrease of the neuronal calcium
channels activity and as a consequence to a reduction of the synaptic hyperexcitability. Recent studies demonstrate that N-type and T-type calcium
channels are both involved in the transmission of painful messages and represent major pharmacological targets of therapeutic interest.
Keywords:
Calcium channels - Neuropathic pain -
α
2-ligands.
* Département de physiologie, Institut de génomique fonctionnelle, CNRS
UMR5203, INSERM U661, université Montpellier I et II, 34094 Montpellier.
DIVERSITÉ FONCTIONNELLE DES CANAUX CALCIQUES
L’électrophysiologie cellulaire, en particulier la technique du
patch clamp,
permet une approche fonctionnelle de l’activité
des canaux calciques par la mesure directe du courant trans-
membranaire transporté par les ions Ca2+ au niveau de nom-
breuses cellules excitables et non excitables.
La classification fonctionnelle des canaux calciques est fondée
sur des critères électrophysiologiques et pharmacologiques suite
à la découverte de ligands et de toxines spécifiques
(1)
. On peut
distinguer deux catégories de canaux calciques en fonction de
leur seuil d’activation. La première catégorie est activée par des
dépolarisations relativement élevées (> – 30 mV) et correspond
aux canaux calciques de type “haut seuil”, ou HVA
(high-voltage-
activated)
. Plusieurs types de canaux HVA ont été identifiés,
dont les canaux de type L. Leur inactivation (fermeture) peut être
très lente, avec pour caractéristique essentielle leur sensibilité
aux “antagonistes calciques” utilisés en thérapeutique cardio-

La Lettre du Pharmacologue - Volume 19 - n° 4 - octobre-novembre-décembre 2005
111
DOSSIER
vasculaire et, en particulier, à la classe des dihydropyridines,
outils pharmacologiques très précieux, comprenant à la fois des
antagonistes et des agonistes. Le type L correspond au courant
entrant lent décrit à l’origine sur les préparations musculaires et
joue un rôle majeur dans les couplages excitation-contraction et
excitation-sécrétion. D’autres courants de type HVA ont ensuite
été décrits sur les cellules nerveuses, avec quelques différences
en termes de seuil d’activation, de voltage-dépendance et de
cinétique d’inactivation, mais qu’on distingue essentiellement
sur des critères pharmacologiques. Les canaux de type N sont
spécifiquement bloqués par une toxine peptidique isolée d’un
escargot marin
(Conus geographicus)
:l’ω-conotoxine GVIA
(ω-cTx). Les canaux de type P et de type Q sont bloqués par un
peptide (ω-Aga-IVA) d’un venin d’araignée
(Agelenopsis aperta)
.
Les canaux de type Q qui s’apparentent au type P présentent
une sensibilité moindre à l’ω-Aga IVA, mais sont bloqués beau-
coup plus spécifiquement par une autre toxine extraite de
Conus
magus
:l’ω-conotoxine MVIIC. Les types N et P/Q sont distri-
bués sur un grand nombre de neurones centraux ou péri-
phériques et, notamment, au niveau présynaptique, où ils
contrôlent la libération des neurotransmetteurs et, en consé-
quence, la transmission synaptique
(1, 2).
Enfin, on définit les
canaux de type R comme ceux qui sont résistants aux toxines
citées précédemment, et leur correspondance fonctionnelle n’est
pas parfaitement établie.
DIVERSITÉ STRUCTURALE DES CANAUX CALCIQUES
VOLTAGE-DÉPENDANTS
Tous les canaux calciques activés par la dépolarisation mem-
branaire possèdent une sous-unité transmembranaire appelée
α1,et la diversité fonctionnelle des canaux calciques tient en
premier lieu à la multiplicité de ces sous-unités canalaires α1
ou Cav(tableau I et figure 1). Chaque sous-unité α1présente
des propriétés fonctionnelles distinctes et une expression
tissulaire précise. Une première famille de gènes code pour
les canaux HVA de type L (Cav1), pour lesquels quatre gènes
Sous-unité canalaire Type de courant Pharmacologie Principale expression tissulaire
CaV1.1 (alias α1S)Type L Dihydropyridines (DHP) et autres Muscles squelettiques
classes d’antagonistes calciques
CaV1.2 (alias α1C)Type L // Ubiquitaire : cœur, muscles lisses, neurones et cellules endocrines…
CaV1.3 (alias α1D)Type L // Neurones, cellules sensorielles (rétine, oreille interne),
cœur (sinus, oreillette)
CaV1.4 (alias α1F)Type L // Rétine, mastocytes
CaV2.1 (alias α1A)Type P/Q ω-agatoxine IVA Système nerveux, jonction neuromusculaire, cellules ß pancréatiques
CaV2.2 (alias α1B)Type N ω-conotoxine GVIA Système nerveux central et périphérique (incluant nocicepteurs)
ω-conotoxine MVIIA
Ziconotide
CaV2.3 (alias α1E)Type R SNX482 Systèmes nerveux et endocrines
CaV3.1 (alias α1G)Type T Mibéfradil Systèmes nerveux et endocrines, cœur
Éthosuximide et forte sensibilité
CaV3.2 (alias α1H)Type T // Systèmes nerveux et endocrines
Nickel Neurones nocicepteurs
CaV3.3 (alias α1I)Type T // Système nerveux
Tableau I. Caractéristiques essentielles des canaux calciques voltage-dépendants.
Figure 1. Structure multimérique des canaux calciques voltage-
dépendants de type haut seuil (HVA). Le complexe canal calcique
est formé de la sous-unité
α
1,qui constitue le pore laissant passer
les ions calcium suite à un changement de conformation induit par
la dépolarisation membranaire. Elle est associée aux sous-unités
régulatrices ß (cytosolique) et
α
2-
δ
.
Noter que
α
2-
δ
est formée de deux protéines associées par un pont
disulfure,
α
2étant extracellulaire et
δ
principalement transmem-
branaire.
Membrane γα
α2
Ca2+
ß
δ

112
La Lettre du Pharmacologue - Volume 19 - n° 4 - octobre-novembre-décembre 2005
DOSSIER
ont été identifiés (Cav1.1, Cav1.2, Cav1.3, Cav1.4). Une
deuxième famille correspond aux canaux HVA de type non-L
ou “neuronaux”, générés par les sous-unités Cav2.1, Cav2.2 et
Cav2.3, qui codent respectivement pour les canaux de
type P/Q, de type N et de type R
(1, 2).
Une troisième famille
correspond aux canaux calciques de type T, représentés par
trois isoformes (Cav3.1, Cav3.2, Cav3.3) activés par de faibles
dépolarisations membranaires
(3).
La pharmacologie des
canaux de type T est pauvre comparativement à celle des autres
canaux calciques. Le mibéfradil a été présenté comme un
antagoniste spécifique des canauxT, mais il affecte également
les canaux calciques de type L ainsi que les canaux neuro-
naux N et P/Q. L’absence de ligands spécifiques a fait que le
rôle physiologique des canaux de type T reste hypothétique,
même si leur implication dans les activités rythmiques neuro-
nale et cardiaque (pacemaker) a été fortement suggérée, ainsi
que dans certains processus physiopathologiques (proliféra-
tion cellulaire, épilepsie, hypertrophie cardiaque…).
Les études structure-fonction dans des modèles d’expression
fonctionnelle ont permis d’identifier sur α1les structures
essentielles
liées à la fonction de ces canaux, comme les struc-
tures impliquées dans le pore (canal), le senseur de voltage ou
encore les sites pharmacologiques.
L’épissage alternatif génère de nombreuses isoformes pour
chacune des sous-unités α1,et il est maintenant établi que ce
mécanisme de régulation transcriptionnelle accroît fortement
la diversité des canaux calciques. Ainsi, les courants de type P
et Q correspondent par exemple à deux variants du gène qui
code pour la sous-unité Cav2.1. La présence d’une valine dans
la boucle intracellulaire qui relie les domaines I et II puis l’in-
sertion de deux amino-acides asparagine et proline (NP) dans
une portion extracellulaire du domaine IV qui réduit la sensi-
bilité du canal vis-à-vis de l’ω-agatoxine IVA permettent de
rendre compte des différences fonctionnelles entre les cou-
rants de type P et Q
(4)
.
LES SOUS-UNITÉS AUXILIAIRES RÉGULATRICES : ß, α2-δET γ
La purification biochimique du canal calcique musculaire
puis les études structure-fonction ont révélé que plusieurs
sous-unités étaient associées aux sous-unité α1de type HVA
(Cav1.x Cav2.x) : les sous-unités ß, α2-δet la sous-unité γ. La
sous-unité γa pendant longtemps été considérée comme asso-
ciée uniquement au canal musculaire (Cav1.1). L’identi-
fication récente de huit isoformes neuronales a relancé le
débat, mais il n’a pas été démontré qu’elles soient partie inté-
grante du complexe formant les canaux calciques neuronaux.
Dans le contexte de cette revue, cette sous-unité ne sera pas
considérée
(5)
.
La sous-unité ß est cytosolique et quatre isoformes ont été
identifiées (ß1 à ß4), ainsi que de nombreux variants d’épis-
sage. Les isoformes ont un domaine conservé d’interaction
(AID) avec les sous-unités α1de type HVA. Les sous-unités ß
favorisent l’adressage membranaire de la sous-unité α1en
masquant sur celle-ci un signal de rétention par le réticulum
sarcoplasmique. Les sous-unités ß modulent les propriétés
biophysiques des canaux HVA avec des caractéristiques spé-
cifiques selon les combinaisons α1-ß. L’association d’une
sous-unité ß augmente l’amplitude du courant calcique et va
interférer avec certaines régulations majeures des canaux cal-
ciques parmi lesquelles la régulation par des récepteurs mem-
branaires couplés aux protéines G (RCPG) comme les récep-
teurs opioïdes
(6)
.
La sous-unité α2-δcorrespond au produit d’un seul gène qui
va, au niveau post-traductionnel, être clivé en peptides α2et δ
liés par un pont disulfure. Quatre gènes ont été identifiés (α2-
δ1 à α2-δ4) ainsi que divers variants d’épissage. La sous-
unité α2est extracellulaire et est considérée comme celle
ayant la fonction régulatrice sur α1. La coexpression d’une
sous-unité α2-δavec une sous-unité α1de type haut seuil
(HVA) induit une augmentation de l’amplitude du courant
calcique pouvant être attribuée à une augmentation de l’adres-
sage membranaire d’α1et/ou à une interaction allostérique
facilitatrice sur l’activité canal d’α1. Les propriétés biophy-
siques du canal calcique sont aussi modifiées, en particulier
sa voltage-dépendance, mais, ici encore, il faut souligner que
ces modulations sont dépendantes non seulement des combi-
naisons d’isoformes entre α1et α2-δ,mais aussi de l’iso-
forme ß associée
(7)
. Cela renforce le concept selon lequel un
canal calcique est un complexe formé de plusieurs sous-unités
et que les combinaisons spécifiques d’isoformes entre la
sous-unité canal α1et les sous-unités régulatrices dans un
tissu donné vont avoir des conséquences fonctionnelles
majeures incluant la modulation de leur pharmacologie.
RÔLE ET FONCTION DES CANAUX DE TYPE N
DANS LA DOULEUR
Les canaux calciques de type N sont codés par la sous-unité
α1B (Cav2.2), qui peut aussi générer plusieurs variants, loca-
lisés principalement au niveau présynaptique. Ces canaux
sont exprimés spécifiquement dans le système nerveux et à
des niveaux très élevés dans la couche superficielle de la
corne dorsale de la moelle épinière (lamina I et II), qui est le
lieu des connexions synaptiques entre les neurones nocicep-
tifs primaires afférents et les voies ascendantes centrales.
L’ouverture des canaux N par les potentiels d’action propagés
via les ganglions rachidiens dorsaux (DRG) conduit à une
entrée du calcium et à la libération des neurotransmetteurs.

La Lettre du Pharmacologue - Volume 19 - n° 4 - octobre-novembre-décembre 2005
113
DOSSIER
Une des voies principales de régulation de l’influx présynap-
tique est l’inhibition de ces canaux N par l’activation des
RCPG, comme par exemple les récepteurs µ aux opioïdes,
une voie de régulation essentielle dans le cadre de la nocicep-
tion. Ainsi, dans les ganglions rachidiens dorsaux (DRG),
l’activation des récepteurs aux opioïdes qui inhibe les
canaux N via les sous-unités ßγdes protéines G
(8)
exerce un
effet antinociceptif. De plus, la souris inactivée pour le
canal N (KO Cav2.2) est moins sensible aux douleurs neuro-
pathiques et inflammatoires sans autre modification phéno-
typique, faisant des canaux N une cible d’intérêt pour le traite-
ment des douleurs chroniques. En ce sens, le peptide synthé-
tique dérivé de la toxine ω-conotoxine MVIIA, le ziconotide,
induit, après injection intrathécale chez l’animal mais aussi
chez l’homme, une forte analgésie sans phénomène de tolé-
rance apparent
(8)
,mais avec certains effets indésirables, en
particulier cardiovasculaires (hypertension). Étant donné son
administration directe au niveau de la moelle épinière, son
utilisation reste limitée au traitement de patients hospitalisés
pour cancer ou pour des douleurs très sévères. Un autre
peptide dérivé de l’ω-conotoxine CVID (AM-336, Amrad
Corporation, Australie) qui bloque les canaux de type N est
aussi en phase 1 chez l’homme, et il semble avoir une fenêtre
thérapeutique plus large que le ziconotide
(9)
. Il est évident
que des industries pharmaceutiques tentent de développer
de petites molécules organiques antagonistes des canaux N
d’administration orale à visée antalgique. Il est à noter qu’un
variant d’épissage des canaux N spécifique des neurones
sensoriels a été identifié et décrit comme très abondant dans
les neurones nociceptifs
(10)
. La perspective de développer
un antalgique qui cible spécifiquement ce variant est intéres-
sante à explorer. La différence de structure porte sur une
portion de la partie carboxy-terminale qui a été impliquée
dans une interaction directe avec le récepteur
(opioid-like)
à la
nociceptine, ces récepteurs exerçant un bloc tonique et consti-
tutionnel des canaux N en l’absence d’agonistes qui est pro-
portionnel à la densité des récepteurs ORL1 exprimés
(11).
Sachant que ces récepteurs sont surexprimés lors d’un traite-
ment par la morphine, les effets de la morphine sur les
canaux N sont d’autant plus atténués, voire supprimés, lors
d’une surexpression des récepteurs ORL1, un mécanisme
pouvant contribuer au développement de la tolérance à la
morphine.
LES CANAUX DE TYPE T COMME CIBLES
DE NOUVEAUX ANALGÉSIQUES ?
Les canaux calciques de type T qui sont impliqués dans l’ex-
citabilité et les activités rythmiques sont de bons candidats
pour jouer un rôle dans la transmission de la douleur.
Plusieurs arguments accréditent cette hypothèse. Leur seuil
d’excitation est bas (< – 40 mV), comme pour les canaux
sodiques TTX-résistants, dont on a démontré un rôle dans la
douleur neuropathique inflammatoire. Ils sont fortement
exprimés dans les neurones DRG et, en particulier, dans les
neurones de petit et moyen diamètre correspondant aux neu-
rones nociceptifs. Une sous-population de neurones DRG
nociceptifs exprime des densités élevées de ce canal, notam-
ment les neurones de la mécano-sensation
(12)
. Les canaux T
sont aussi exprimés dans la couche superficielle de la corne
dorsale de la moelle épinière [lamina I et II]
(13)
.
Parallèlement à cette forte expression, l’activité des canaux T
des neurones DRG contribue à la genèse de potentiels excita-
teurs dans les neurones postsynaptiques de la corne antérieu-
re de la moelle épinière et à la potentialisation à long terme
(LTP) de ces synapses
(14)
.
Tous ces éléments rendaient probable le rôle des canaux T
dans la transmission des informations nociceptives, mais
l’absence de pharmacologie spécifique a limité les études
fonctionnelles. Seul élément, l’administration systémique
de mibéfradil, une molécule considérée comme relativement
sélective des canaux T, exerce un effet antinociceptif dans
les douleurs neuropathiques
(15).
Similairement, l’éthosuxi-
mide, utilisé pour ses propriétés antiépileptiques et qui est
décrit comme un inhibiteur des canaux T, a un effet anti-
douleur
(16).
Cependant, ces molécules affectant d’autres
types de canaux qui peuvent aussi être impliqués dans la
transmission de la douleur, ces données n’étaient pas
concluantes.
Les canaux de type T sont codés par trois gènes correspon-
dant aux isoformes Cav3.1, Cav3.2, Cav3.3 (alias α1G,α1H,
α1I). Ces isoformes ont un profil d’expression particulier avec
une forte expression de l’isoforme Cav3.1 dans le thalamus,
alors qu’elle est absente dans les DRG qui expriment quasi
exclusivement l’isoforme Cav3.2 et, à faible niveau, Cav3.3
(8)
. Les travaux de notre laboratoire ont démontré très récem-
ment, par une approche antisens in vivo, le rôle des canaux T
dans la douleur. L’injection intrathécale d’antisens dirigés
spécifiquement contre chacune des isoformes codant pour les
canaux T a montré le rôle spécifique de l’isoforme Cav3.2
dans les douleurs mécaniques et thermiques
(17)
[figure 2].
Un traitement par des antisens durant quatre jours induit une
réponse antinociceptive comparable à celle observée en
présence de morphine, mais qui se maintient pendant plu-
sieurs jours après l’arrêt du traitement. L’effet est encore plus
prononcé sur le modèle neuropathique de ligature du nerf
sciatique (CCI). Ces résultats indiquent que le canal de type T
généré par l’isoforme Cav3.2 représente une cible d’intérêt
pour le développement de nouvelles molécules antinoci-
ceptives.

114
La Lettre du Pharmacologue - Volume 19 - n° 4 - octobre-novembre-décembre 2005
DOSSIER
LES LIGANDS DE LA SOUS-UNITÉ α2-δDES CANAUX
CALCIQUES DANS LE TRAITEMENT DES DOULEURS
NEUROPATHIQUES
Il est admis qu’une sous unité α2-δest associée à tous les
types de canaux calciques de type haut seuil. Quatre iso-
formes ont été identifiées, et la coexpression d’une sous-unité
α2-δavec une sous-unité α1de type haut seuil dans un modèle
d’expression induit une augmentation de l’influx calcique
attribuée soit à une interaction structurale facilitatrice exercée
sur α1,soit à une augmentation de la densité des canaux fonc-
tionnels par un mécanisme favorisant l’adressage membranaire
de la sous-unité α1
(18)
. Une nouvelle classe de molécules
dont le premier représentant a été la gabapentine induit une
analgésie dans les douleurs neuropathiques sur des modèles
animaux et chez l’homme
(19)
. La prégabaline a un profil
pharmacologique similaire, mais une plus grande efficacité
dans les modèles précliniques de douleur et d’épilepsie
(20)
.
Le mécanisme d’action de ces molécules n’a pas été claire-
ment identifié, mais un élément essentiel révélé par les études
de liaison est qu’elles se lient à la sous-unité α2-δ,d’où le
terme “α2-ligands”
(21)
,et en particulier à la sous-unité
α2-δ1,pour laquelle l’affinité est la plus élevée par rapport à
la sous-unité α2-δ2
(18)
. La fixation sur α2-δ1est nécessaire
pour observer les effets antinociceptifs qui sont supprimés
chez la souris exprimant une sous-unité α2-δ1avec une muta-
tion ponctuelle qui empêche la fixation de la prégabaline
(9).
Ces deux isoformes, dont l’expression est relativement ubi-
quitaire, mais avec un profil d’expression variable selon les
tissus, sont fortement exprimées dans les neurones nociceptifs
des ganglions rachidiens
(22)
. Une observation importante est
la très forte surexpression de la sous-unité α2-δ1du côté ipsi-
latéral après ligature du nerf dans le modèle de rat neuropa-
thique de Kim et Chung
(23).
Cette augmentation d’α2-δ1
précède l’allodynie tactile ipsilatérale qui peut être atténuée
par l’infusion intrathécale de gabapentine. De plus, cette sur-
expression d’α2-δ1dans un modèle neuropathique disparaît
progressivement parallèlement à l’allodynie tactile après
plusieurs semaines. Ces données établissent donc une corres-
pondance claire et temporelle entre le niveau d’expression
d’α2-δ1et l’apparition ou la disparition de l’allodynie. De
plus, cette surexpression d’α2-δ1n’est pas observée lors de
la rhizotomie dorsale (section du nerf entre le corps cellulaire
du DRG et la corne dorsale), démontrant que l’expression
d’α2-δ1se produit dans les DRG et est régulée par des
facteurs périphériques. En fait, la lésion périphérique par liga-
ture du nerf sciatique (modèle CCI) induit aussi une surex-
pression d’α2-δ1dans la corne dorsale de la moelle épinière
qui est corrélée à l’allodynie tactile neuropathique. La rhizo-
tomie dorsale inhibe la surexpression d’α2-δ1induite par la
lésion périphérique, ce qui indique que cette surexpression est
principalement présynaptique. La surexpression de la sous-
unité α2-δ1se produirait dans le DRG suite à la lésion, puis
celle-ci migrerait par transport axonal dans la corne dorsale,
où elle régulerait la neurotransmission
(24)
. En accord avec
ces données, l’injection intrathécale d’antisens sur ce modèle
neuropathique diminue fortement à la fois la surexpression
d’α2-δinduite par la lésion dans les DRG et dans la corne
dorsale et l’allodynie tactile associée. L’ensemble de ces
résultats suggère que la surexpression d’α2-δconstitue un
élément clé de la plasticité neuronale liée au développement
de l’allodynie suite à une lésion nerveuse périphérique. Des
études de cryomicroscopie ont montré la proximité entre α2
et α1dans la membrane
(25).
Il est probable que cette sous-
unité joue un rôle critique dans l’assemblage et l’expression
des canaux calciques neuronaux, sa surexpression pouvant
donc augmenter le nombre et/ou l’activité des canaux
Figure 2. Les canaux calciques T périphériques ont un rôle impor-
tant dans la transmission des messages douloureux. Des antisens
contre une seule isoforme (Cav3.2) administrés par voie intrathé-
cale induisent une très forte analgésie maintenue pendant plusieurs
jours sur un modèle de douleur neuropathique chez le rat (test Ran-
dall et Selito). Parallèlement, le courant calcique T enregistré dans
les neurones DRG de petit et moyen diamètre (nocicepteurs) à par-
tir de ces animaux traités par antisens est fortement réduit.
800
700
600
500
400
300
200
100
Seuil de douleur (g)
– 7
Avant
chirurgie
Après
chirurgie
Jours
Douleur mécanique (test de pression de la patte)
04567891011 12
*
*
*
*
Contrôle
AS Cav3.2
*
*
Injections antisens
10
8
6
4
2
0
Courants de type T (pA/pF)
Contrôle As Cav3.2
Contrôle AS Cav3.2
Courants de type T à J+4
(n = 53)
(n = 43)
20 ms
200pA
 6
7
6
7
1
/
7
100%