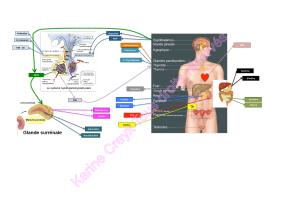
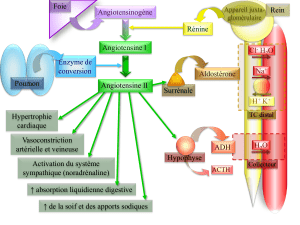
Rôles physiopathologiques de l`aldostérone

FLAMMARION
MÉDECINE
-
SCIENCES
—
ACTUALITÉS
NÉPHROLOGIQUES
2005
(www.medecine.flammarion.com)
RÔLES PHYSIOPATHOLOGIQUES DE L’ALDOSTÉRONE :
APPORT DES MODÈLES ANIMAUX
par
Y. SAINTE-MARIE, A. OUVRARD-PASCAUD, F. JAISSER*
Si la définition d’hormone minéralocorticoïde, c’est-à-dire modulant la réab-
sorption du sodium dans les cellules rénales, est toujours d’actualité pour l’aldos-
térone, ces dernières années ont vu l’émergence d’une série de problématiques
soulevées par la mise en évidence de nouvelles cellules cibles, épithéliales ou non,
où le rôle de l’hormone reste mal connu. Son implication en pathologie s’est élargie
de façon spectaculaire (fibrose et insuffisance cardiaque, troubles de la mémoire,
maladies génétiques affectant la pression artérielle), renouvelant l’intérêt de l’ana-
lyse de son mécanisme d’action dans des perspectives pathogéniques multiples, et
stimulant la recherche de modèles cellulaires et animaux susceptibles de faire pro-
gresser les connaissances, ou encore d’offrir des outils pour tester de nouvelles
approches thérapeutiques.
ALDOSTÉRONE/MINÉRALOCORTICOÏDES :
IMPLICATION EN PHYSIOPATHOLOGIE
CARDIOVASCULAIRE ET RÉNALE
L’hormone minéralocorticoïde aldostérone participe au contrôle de la réabsorp-
tion rénale de sodium jouant ainsi un rôle majeur dans la régulation de la volémie
et de la pression artérielle comme l’ont montré les patients avec un syndrome de
perte de sel lié à une mutation inactivante du récepteur de l’aldostérone [1]. L’hor-
mone exerce ses effets dans le néphron distal après liaison au récepteur minéralo-
corticoïde (RM), un récepteur nucléaire qui agit comme un facteur de transcription
dépendant des ligands [2].
* INSERM Avenir IFR 52, Collège de France, Paris.

182 Y. SAINTE-MARIE ET COLL.
Au cours des dix dernières années, il a été démontré que le RM est également
exprimé dans des cellules non épithéliales comme les cardiomyocytes ou les cel-
lules endothéliales et musculaires lisses des vaisseaux. L’expression du RM dans
les cardiomyocytes a ainsi été montrée chez le lapin, le rat, la souris ou l’homme
[3, 4, 5]. L’existence d’un système paracrine a été proposé : la voie de synthèse
de l’aldostérone (CYP11B2) a été mise en évidence dans le cœur et la synthèse
locale d’aldostérone a été rapportée [6], en particulier chez l’homme, en cas
d’insuffisance cardiaque [7].
Le rôle physiopathologique du RM et de l’aldostérone dans ces cibles non clas-
siques reste à déterminer [8]. De nombreuses données suggèrent que l’aldostérone
agit comme un médiateur dans certaines pathologies dégénératives comme l’insuf-
fisance rénale chronique ou le remodelage artériel ou myocardique, indépendam-
ment ou en synergie des effets connus de l’angiotensine II [8, 9]. La vasculopathie
liée à l’aldostérone pourrait être une phase clé de plusieurs pathologies cardio-
vasculaires, spécialement dans l’insuffisance cardiaque où on peut observer une
hyperaldostéronémie plus ou moins importante. Les effets vasculaires de l’aldo-
stérone pourraient ainsi directement participer à l’hypertension artérielle [10] et
au remodelage myocardique secondaire aux altérations des vaisseaux coronariens
[11, 12, 13]. Dans le modèle animal du rat aldostérone-sel et chez l’homme, le
facteur déclenchant de la cardiopathie pourrait être une vasculopathie induite par
l’aldostérone, via l’activation du RM. Les antagonistes du RM atténuent également
le remodelage artériel associé à diverses vasculopathies (athérosclérose, diabète)
[14, 15].
ALDOSTÉRONE/MINÉRALOCORTICOÏDES :
UNE NOTION ÉMERGENTE DES PATHOLOGIES
DÉGÉNÉRATIVES CHEZ L’HOMME
L’implication clinique du rôle physiopathologique du couple aldostérone/RM
dans les pathologies dégénératives a été étudiée dans plusieurs essais cliniques
chez l’homme. L’étude RALES a clairement démontré que la spironolactone, un
inhibiteur du RM, chez des patients en insuffisance cardiaque sévère, en complé-
ment d’un traitement classique utilisant les inhibiteurs de l’enzyme de conversion
de l’angiotensinogène diminue le risque de mortalité de 30 p. 100 [16]. Récem-
ment, l’essai EPHESUS utilisant un antagoniste plus spécifique, l’éplérenone, a
également montré un effet bénéfique dans des cardiopathies ischémiques [17].
Les effets dans RALES et EPHESUS sont dus pour moitié à une diminution des
morts subites [16, 17]. Jusqu’à présent, les mécanismes et cibles cellulaires impli-
qués dans ces effets n’ont pas clairement été identifiés. Il faut noter que l’activa-
tion du RM chez l’homme a des conséquences électrophysiologiques majeures.
La fludrocortisone, minéralocorticoïde synthétique, exacerbe [18, 19, 20] les
paramètres électrophysiologiques cliniques de risque d’arythmie tel que la dis-
persion de l’interval QT, alors que la spironolactone les améliore. La spironolac-
tone augmente la variabilité du rythme sinusal et diminue les extrasystoles
ventriculaires et les épisodes de tachycardie ventriculaire des patients en insuffi-
sance cardiaque, au repos ou à l’effort [21]. Des données expérimentales obtenues
sur des cardiomyocytes en culture primaire indiquent que l’aldostérone par elle-

RÔLES PHYSIOPATHOLOGIQUES DE L’ALDOSTÉRONE 183
même induit un remodelage ionique (augmentation du courant calcique, diminu-
tion de certains courant potassique, allongement de la durée du potentiel d’action)
[22, 23].
Dans le rein, l’aldostérone a récemment été proposée comme acteur de la pro-
gression de l’insuffisance rénale, indépendamment de ses effets hémodynamiques
traditionnels. Ce concept constitue un changement par rapport à la notion classique
de l’aldostérone agissant comme hormone de contrôle de la balance hydro-sodée
[24, 25]. Ainsi, la spironolactone réduit la protéinurie chez des patients en insuf-
fisance rénale chronique [26] ou présentant une néphropathie diabétique [27]. La
participation vasculaire à l’action délétère de l’aldostérone a été évoquée dans ces
effets bénéfiques, et clairement identifiée dans le modèle pharmacologique du rat
aldostérone-sel ou le modèle génétique du rat SHR pour les altérations rénales qui
leur sont associés [11, 28, 29, 30].
En conclusion, l’aldostérone peut être considérée, chez l’homme, comme un
facteur de risque important dans les pathologies cardiovasculaires et rénales.
SIGNALISATION DE L’ALDOSTÉRONE DANS LE CŒUR
La signalisation du couple aldostérone/RM est difficile à analyser car les inte-
ractions moléculaires et fonctionnelles avec les hormones glucocorticoïdes et leur
récepteur sont complexes. Il est important de souligner que : 1) le RM lie avec des
affinités comparables l’aldostérone et les glucocorticoïdes, ces derniers pouvant
être inactivés par l’enzyme 11β-hydroxystéroïde déshydrogénase de type II
(HSD2), protégeant ainsi le RM d’une occupation illicite par les glucocorticoïdes ;
2) l’aldostérone à forte concentration peut occuper le récepteur glucocorticoïde
(RG) ; 3) le RM hétérodimérise avec le RG, les hétérodimères ayant des propriétés
de transactivation différentes des homodimères. Il est donc souvent difficile de
dissocier des effets et conséquences moléculaires propres à l’aldostérone et non
aux glucocorticoïdes.
La nature moléculaire des protéines régulées par l’aldostérone de façon précoce
ou tardive dans le rein fait actuellement l’objet de recherches intenses. Plusieurs
gènes sont actuellement reconnus comme des cibles de l’aldostérone dans le canal
collecteur rénal tels que des protéines de transport (ENaC, ROMK, sous-unités α1
de la Na, K-ATPase) ou des gènes impliqués dans la signalisation cellulaire
(SGK, GILZ ou NDRG2) [31-35]. Toutefois, la difficulté d’accès aux cellules
cibles rénales (représentant moins de 1 p. 100 des cellules rénales) et la com-
plexité du mode d’action de l’aldostérone rendent ces études difficiles. La signa-
lisation de l’aldostérone dans le cœur est encore moins bien définie. Les études
menées sur des cardiomyocytes isolés ou dans le modèle pharmacologique aldo-
stérone-sel suggèrent que sont impliqués le système rénine-angiotensine local
ainsi que l’endothéline, l’oxyde nitrique, les anions superoxydes et l’inflamma-
tion [36, 37]. La spécificité de l’aldostérone et des glucocorticoïdes, ainsi que
celles du RM et du RG dans la physiopathologie cardiaque doivent être analysées
avec précaution pour comprendre les actions cardiovasculaires des hormones
minéralocorticoïdes [38]. En effet, chez l’homme, la souris et le rat, la 11β-HSD2
ne serait pas exprimée dans les cardiomyocytes et la faible activité cardiaque de
l’enzyme serait plus probablement d’origine vasculaire [39, 40]. Ceci suggère que

184 Y. SAINTE-MARIE ET COLL.
le ligand prépondérant du RM cardiaque est de type glucocorticoïde (sauf en cas
d’hyperaldostéronémie importante) alors que l’endothélium doit être considéré
comme un tissu cible classique, au même titre que le canal collecteur rénal, car
l’expression de la 11HSD2 y assure la spécificité aldo/gluco requise. D’après des
travaux réalisés sur des cultures néonatales de cardiomyocytes, il semble que le
complexe RG-aldostérone soit fonctionnellement actif et permette de stimuler la
transcription de gènes cibles comme la kinase Sgk (serum and glucocorticoid
activated kinase) [5]. Des recherches complémentaires sont donc indispensables
pour déterminer plus précisément la nature des liaisons hormone/récepteur dans
le cœur.
RÔLES PHYSIOPATHOLOGIQUES DE L’ALDOSTÉRONE
ET DU RM : INTÉRÊTS DES MODÈLES ANIMAUX
Modèles pharmacologiques et pathologiques
Le modèle du rat dit aldostérone/sel est couramment utilisé pour étudier le rôle
physiopathologique de l’aldostérone [41]. Dans ce modèle complexe, les animaux
sont uninephrectomisés, infusés avec une dose robuste d’aldostérone (0,75 μg/h),
et reçoivent un excès de sel dans l’eau de boisson (1 p. 100 NaCl ; 0,3 p. 100 KCl).
Les animaux développent : (a) une hypertension artérielle (de 130 à 190 mmHg)
et une hypertrophie du ventricule gauche (VG) ; (b) une fibrose myocardique
interstitielle et périvasculaire. Le traitement de ces animaux avec une dose sub-
hypotensive de spironolactone prévient l’apparition d’une fibrose, indépendam-
ment d’un effet sur l’hypertrophie du VG [42]. Il n’existerait donc pas de relation
directe entre pression systolique, hypertrophie et fibrose, d’autant que le ventricule
droit non hypertrophié et ne subissant pas les mêmes contraintes hémodynamiques
que le VG, présente une fibrose identique. Autrement dit, la fibrose cardiaque
serait ici d’origine hormonale, liée à l’hyperaldostéronémie mais également au sel.
En effet, des contrôles uninephrectomisés, recevant exclusivement soit un excès
de sel, soit une infusion d’aldostérone, ne présentent pas de fibrose [43]. Dans tous
les cas, un infiltrat inflammatoire accompagne le développement d’une fibrose
périvasculaire, suivi par la mise en place d’une fibrose interstitielle. Des travaux
récents précisent en effet que l’hyperaldostéronisme serait primairement associé à
une activation de cellules immunitaires circulantes, à l’origine d’un stress oxydatif
responsable de l’apparition d’un phénotype cardiaque pro-inflammatoire et fibro-
génique [44].
Le modèle des rats infarcis a également été utilisé pour étudier les effets car-
diaques de l’aldostérone. Suite à l’ischémie, les animaux déclenchent des méca-
nismes adaptatifs de remodelage myocardique, processus indispensable pouvant
néanmoins engendrer des effets délétères comme le développement d’une fibrose
excessive, d’une hypertrophie et d’une dilatation susceptible de mener à une insuf-
fisance cardiaque. Si l’administration d’éplérénone n’affecte pas le processus répa-
ratif de cicatrisation, elle s’accompagne d’un effet protecteur évident concernant
la fibrose réactive dans les parties viables du myocarde. De plus, l’antagonisme
du RM améliorerait progressivement la compliance cardiaque post-infarctus
(mesurée sur des cœurs isolés et perfusés) [45]. L’intérêt de ce modèle repose sur

RÔLES PHYSIOPATHOLOGIQUES DE L’ALDOSTÉRONE 185
l’absence d’apport externe d’aldostérone et de sel ; les effets délétères observés
sont donc directement reliés à une régulation hormonale endogène en réponse à
un état pathologique.
Modèles génétiquement modifiés
Plusieurs travaux remettent en cause l’idée selon laquelle de fortes concentra-
tions d’aldostérone plasmatique suffiraient à induire un phénotype cardiaque.
Wang et al. ont utilisé une lignée de souris transgéniques présentant un pseudo-
hypoaldostéronisme de type I (PHA-I) partiellement compensé qui s’accompagne
d’une hyperaldostéronémie notable (élévation d’un facteur 6) [46]. Soumis à un
régime en sel normal, les animaux ne présentent aucun signe d’hypertrophie, de
remodelage ou de fibrose cardiaque.
Les souris transgéniques P1-hRM générées par Le Menuet et al. expriment le
RM humain (hRM), sous contrôle du promoteur P1 permettant de mimer la distri-
bution tissulaire du RM endogène [47]. Les souris P1-hRM ne développent pas
d’hypertension artérielle. Un phénotype rénal comprend une dilatation tubulaire et
une diminution de 30 p. 100 de l’excrétion urinaire du K+ sans modification de la
kaliémie, suggérant une augmentation de la réabsorption de K+ pour pallier une
déplétion chronique. Au plan cardiaque, le phénotype se caractérise par une car-
diomyopathie dilatée de moyenne amplitude, l’absence de fibrose, une fréquence
cardiaque augmentée et des troubles du rythme observés dans 15 p. 100 des cas.
Les modèles animaux présentant une inactivation non ciblée du gène RM
(entraînant une mortalité périnatale liée à une perte rénale de sel [48]), une sur-
expression constitutive globale du RM [47], ou les modèles pharmacologiques
(manipulant les ligands ou antagonistes du RM et RG) sont complexes à analyser.
En effet, les effets multiples de l’aldostérone et des glucocorticoïdes, ainsi que la
distribution tissulaire large de leur récepteurs spécifiques rendent difficile la recon-
naissance de leur contribution à la physiopathologie cellulaire et/ou tissulaire. Les
modèles transgéniques conditionnels et/ou inductibles permettent un contrôle spatio-
temporel de l’expression du RM ou RG spécifiquement dans des cibles sélection-
nées, mais également un contrôle précis de l’expression au cours du temps [49].
L’approche ciblée évite la possibilité d’effets secondaires dus aux altérations de
l’homéostasie ionique ou aux perturbations globales induites par l’aldostérone ou
les glucocorticoïdes et permet de se focaliser sur le rôle spécifique de leurs récep-
teurs dans les cardiomyocytes ou les vaisseaux [50]. Des modèles ciblés ont donc
été développés, et utilisent tous le promoteur de la chaîne lourde de l’alpha myo-
sine, spécifique des cardiomyocytes, pour diriger l’expression d’un transgène
d’intérêt dans ces seules cellules.
La sous-expression du RM endogène de souris, spécifiquement dans les cardio-
myocytes, a été obtenue à l’aide d’un modèle conditionnel par expression d’un
ARN antisens complémentaire du transcrit endogène [51]. La diminution de moitié
de l’expression du RM est associée au développement d’une cardiomyopathie
hypokinétique dilatée sévère accompagnée de fibrose interstitielle. Ce modèle pré-
sente un phénotype inattendu en comparaison de l’efficacité du traitement par un
antagoniste du RM (spironolactone ou éplérenone) dans l’insuffisance cardiaque
expérimentale (aldostérone/sel, ischémie cardiaque) ou humaine. Un phénotype à
l’état basal n’était pas attendu mais plutôt une protection des souris lors de l’induc-
tion de pathologies. Ce modèle suggère donc que l’implication du RM dans une
 6
7
8
9
10
6
7
8
9
10
1
/
10
100%