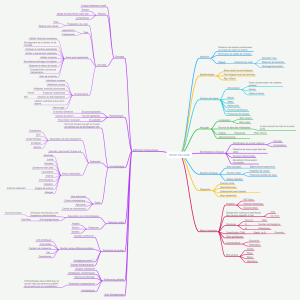
Sommaire - Faculté de Médecine et de Pharmacie Fès

Sommaire

Sommaire :
I) Introduction ……………………………………………………………………… 1
II) Historique………………………………………………………….……………… 3
III) Epidémiologie………………………………………………………………… 4
IV) Embryologie ………………………………………………………………… 5
1) Développement du colon et rectum……………………………..……… 5
2) Développement du système nerveux…………………………………… 5
V) Rappel anatomique……………………………………………………………… 8
1) Le colon……………………………………………………………….……… 8
1-1) La disposition générale…………………………………………… 8
1-2) La division anatomochirurgicale………………………………… 8
1-3) Moyens de fixité et principaux rapports……………………… 9
1-4) La vascularisation artérielle……………………………….……… 10
1-5) La vascularisation veineuse……………………………….……… 10
1-6) Le drainage lymphatique………………………………….……… 11
1-7) L’innervation colique……………………………………….……… 12
2) Le rectum……………………………………………………………….…… 12
2-1) la disposition générale………………………………………..…… 12
2-2) principaux rapports et moyens de fixité…………………..…… 13
2-3) vascularisation artérielle…………………………………………… 13
2-4) les veines du rectum………………………………………………… 14
2-5) les nerfs du rectum………………………………………..………… 15
VI) Rappel anatomopathologique……………………………………….…… 16
1) Les lésions macroscopiques …………………………………………… 16
2) Les lésions macroscopiques …………………………………………… 17
VII) les hypothèses étiologiques …………………………………………………… 19
1) Hypothèse vasculaire…………………………………..………………… 19

2) Hypothèse virale………………………………………………………… 19
3) Hypothèse de neuromédiateurs……………………………………… 19
4) Hypothèse auto-immune……………………………………………… 19
5) Hypothèse génétique…………………………………………………… 19
VIII) Le diagnostic……………………………………………………………..……… 22
1) La clinique …………………………………………………………………… 22
2) Les explorations radiologiques………………………………………… 23
2-1) abdomen sans préparation……………………………………… 23
2-2) le lavement opaque………………………………………………… 23
3) Les explorations non radiologiques…………………………………… 25
3-1) la rectomanométrie………………………………………………… 25
3-2) l’anatomie pathologie………………………………….…………… 26
IX) Les formes cliniques…………………………………………………………… 27
1) Les formes cliniques……………………………………………………… 27
2) Les formes selon l’âge…………………………………………………… 27
3) Les formes compliquées……………………………………….………… 28
X) Les pathologies associées……………………………………………………. 29
1) La trisomie………………………………………………………….………. 29
2) Anomalies urinaires………………………………………………………. 29
3) Anomalies digestives……………………………………………………… 29
4) Autres anomalies associées……………………………………………… 30
XI) Le diagnostic différentiel……………………………………………………… 31
1) Occlusions organiques…………………………………………………… 31
2) Occlusions fonctionnelles……………………………………………..… 31
3) Pseudo obstruction intestinale chronique (POIC)…………………… 31
4) Autres causes de constipation à la période néonatale…………… 32
XII) Le traitement………………………………………………………………..…… 34

1) Buts et principes…………………………………………………………… 34
2) Le traitement d’attente…………………………………………………… 34
3) Le traitement radical……………………………………………………… 38
XIII) Les indications……………………………………………………………..…… 55
1) Forme habituelle recto-sigmoidienne du nouveau né……….……. 55
2) Formes longues de la maladie d’hirschsprung……………..……….. 55
3) Les formes coliques totales……………………………………..………. 55
4) Les formes courtes……………………………………………..…………. 56
XIV) Résultats et complications…………………………………………….…….. 56
1) La mortalité………………………………………………………..………. 56
2) Les complications post opératoires précoces……………..……….. 56
3) L’évolution à long terme………………………………………………… 57
XV) Patients, méthodes et résultats……………………………………………… 59
1) Matériel d’étude…………………………………………………………… 59
2) Méthodes d’études………………………………………….……………. 59
XVI) Résultats et discussion……………………………………………………….. 65
1) Age au moment du diagnostic………………………………….……… 65
2) Sexe…………………………………………………………………….……. 69
3) L’origine géographique…………………………………………….……. 71
4) Antécédents et pathologies associées………………………….…….. 71
5) Les aspects cliniques………………………………………….………….. 72
6) Paraclinique…………………………………………………….………….. 77
7) Le traitement……………………………………………………..………… 82
Conclusion……………………………………………………………………………….…. 87
Résumé………………………………………………………………………………………. 89
Bibliographie……………………………………………………………….………………. 93

Glossaire
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
1
/
106
100%