Hyperplasies Congénitales des Surrénales

HYPERPLASIE CONGENITALE
DES SURRENALES
Dr Claire-Lise Gay – Pr Pierre Chatelain
Centre de Référence des Maladies Rares du Développement
et de la Différenciation Sexuelles (DSD) – Site de Lyon
Coordinateur Pr Pierre Chatelain

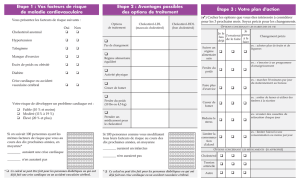
Définition
Ensemble de maladies autosomiques récessives
liées à déficit de la biosynthèse du cortisol
5 enzymes assurent transformation du cholesterol
en cortisol: chaque étape peut être affectée
2 groupes: ceux n’affectant que la biosynthèse
surrénale (21 OHase, 11β-OHase, aldosynthase) et
ceux affectant aussi les gonades( 3β-HSD, 17α-
OHase)
+ 90% cas =déficit en 21-OH ase
1 NN / 16 000, hétérozygote pour mutation sévère:
1/50 à 1/60 (plus fréquent à la Réunion: 1/400)

Biosynthèse des hormones
surrénaliennes
Minéralocorticoïdes Glucocorticoïdes Androgènes
18-OH-Corticostérone
Aldostérone
Pregnénolone
Progestérone
DOC
Cholestérol
(mitochondrie
)
Cholestérol
(cytosol
)
Corticostérone
Corticostérone
Cholestérol
(
cytosol
)
Cholestérol
(mitochondrie)
Pregnénolone
Progestérone
DOC
StAR
P450scc
3ß-HSD
P450c21
21-hydroxylase
P450c18
11ß-hydroxylase
18-hydroxylase
18-déshydrogénase
17-OH-pregnénolone
17
-
OH
-
progéstérone
11-désoxycortisol
Cortisol
P450c21
21-hydroxylase
3ß-HSD
DHA
∆4 androstenedione
3ß-HSD
17ß-HSD
5 α
réductase
Testostérone
DHT
P450c11
11ß-
h
ydroxylase
P450c17
17 α-hydroxylase/17,20-
lyase

Biosynthèse des hormones
surrénaliennes
Minéralocorticoïdes Glucocorticoïdes Androgènes
18-OH-Corticostérone
Aldostérone
Pregnénolone
Progestérone
DOC
Cholestérol
(mitochondrie
)
Cholestérol
(cytosol
)
Corticostérone
Corticostérone
Cholestérol
(
cytosol
)
Cholestérol
(mitochondrie)
Pregnénolone
Progestérone
DOC
StAR
P450scc
3ß-HSD
P450c21
21-hydroxylase
P450c18
11ß-hydroxylase
18-hydroxylase
18-déshydrogénase
17-OH-pregnénolone
17
-
OH
-
progéstérone
11-désoxycortisol
Cortisol
P450c21
21-hydroxylase
3ß-HSD
DHA
∆4 androstenedione
3ß-HSD
17ß-HSD
5 α
réductase
Testostérone
DHT
P450c11
11ß-
h
ydroxylase
P450c17
17 α-hydroxylase/17,20-
lyase

Biosynthèse des hormones
surrénaliennes
Minéralocorticoïdes Glucocorticoïdes Androgènes
18-OH-Corticostérone
Aldostérone
Pregnénolone
Progestérone
DOC
Cholestérol
(mitochondrie
)
Cholestérol
(cytosol
)
Corticostérone
Corticostérone
Cholestérol
(
cytosol
)
Cholestérol
(mitochondrie)
Pregnénolone
Progestérone
DOC
StAR
P450scc
3ß-HSD
P450c21
21-hydroxylase
P450c18
11ß-hydroxylase
18-hydroxylase
18-déshydrogénase
17-OH-pregnénolone
17
-
OH
-
progéstérone
11-désoxycortisol
Cortisol
P450c21
21-hydroxylase
3ß-HSD
DHA
∆4 androstenedione
3ß-HSD
17ß-HSD
5 α
réductase
Testostérone
DHT
P450c11
11ß-
h
ydroxylase
P450c17
17 α-hydroxylase/17,20-
lyase
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
1
/
27
100%