Aide numérique à l`identification bactérienne

pratique quotidienne
Aide numérique à l’identification bactérienne :
définition d’un risque d’erreur alpha
B. Van Oystaeyen
Haute école Charleroi-Europe,
site de l’ISC-Fleurus,
Fleurus, Belgique
Article reçu le 8 septembre 2005,
accepté le 4 novembre 2005
Résumé.Le mode de calcul habituellement utilisé en support de l’identifica-
tion bactérienne se révèle efficace dans son rôle premier, qui est de proposer le
nom des bactéries plausibles, mais il échoue dans la définition d’un niveau de
confiance qui puisse être associé à l’éventuelle décision. Pour corriger ce
défaut, l’article propose de calculer l’ensemble des distributions de probabilité
de réponse pour chaque bactérie. Ce calcul utilise la méthode dite de Monte-
Carlo et doit être effectué une fois pour toutes sur une galerie donnée. Sur
chacune de ces distributions, il est aisé de repérer un niveau de risque d’erreur
de première espèce, ou risque d’erreur alpha. L’approche théorique proposée
évite par ailleurs la comparaison de scores d’identification, comparaison qui
pose question dès lors que les différentes bactéries présentent des sensibilités
très différentes aux tests biochimiques.
Mots clés :bactériologie, probabilité, identification, niveau de confiance
Abstract.The numerical method commonly used as an help to the bacterian
identification appears quite efficient in its first role, i.e. to propose the name of
the plausible bacteria, but it fails to define a confidence level to be assiociated
to the final decision. To correct this, this paper propose to calculate the res-
ponse probability distribution for each possible bacteria. The calculation uses
the so-called “Monte-Carlo” method and has to be performed once for all on a
given set of biochemichal tests. For each calculated ditribution, it is easy to find
a first kind error risk, or a-error risk. The proposed theoretical approach
moreover avoid the comparison of “identification scores”, which is always
questionable so far the various bacteria present very different sensitivities to
the biochemical tests.
Key words:bacteriology, probability, identification, confidence level
Pour aborder le problème de l’aide à l’identification des
micro-organismes par méthode numérique, nous suppose-
rons un ensemble de n tests biochimiques s’appliquant à
m bactéries. L’information de base se présente d’habitude
sous la forme d’une matrice de nombres de m lignes et n
colonnes. Le nombre A
ij
affiché à la ligne i, colonne j est
la probabilité pour la i
e
bactérie de répondre positivement
au j
e
test.
Pour identifier un micro-organisme inconnu, connaissant
sa réponse à l’ensemble des tests, les nombres A
ij
sont
combinés selon une procédure que nous rappellerons
ci-dessous et que nous nommerons « approche usuelle »
[1-4]. Ainsi que nous l’avons souligné dans un précédent
article, et ainsi que nous le soulignerons à nouveau,
l’approche usuelle présente un évident défaut de nature
conceptuelle qui trouve son origine dans les formules
mathématiques utilisées et dans la manière de les interpré-
ter. Les praticiens ont depuis longtemps tenté de solution-
ner ce problème, en introduisant à l’appui du calcul de
base des formules auxiliaires [5-7] telles que le biotype. Il
est important de souligner que l’ensemble de la procédure,
faite du calcul de base épaulé par de l’information auxi-
liaire, se révèle de fait efficace et conduit pour l’essentiel à
des identifications dépourvues d’ambiguïté, principale-
ment bien entendu dans les nombreux cas où un candidat
unique émerge du calcul. Cependant, outre le fait qu’elle
abc
Ann Biol Clin 2006 ; 64 (1) : 83-9
Ann Biol Clin, vol. 64, n° 1, janvier-février 2006 83
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

demeure inélégante, l’approche usuelle pose tout simple-
ment question dès qu’il s’agit de discuter des cas où plu-
sieurs candidats à l’identification sont en compétition.
S’agissant de corriger ce défaut, des méthodes alternatives
telles que le biotype sont certainement orientées dans la
bonne direction, mais aucune n’a jamais atteint le statut de
voie d’identification autonome. La raison en est leur inca-
pacité à définir des valeurs seuil telles que le très classique
« risque de première espèce », ou risque ad’identification,
c’est-à-dire des valeurs qui mesurent clairement un niveau
de confiance minimum lié à la décision finale. Le but de
cet article est de montrer que de telles valeurs seuil peu-
vent être obtenues grâce à la puissance de calcul devenue
très commune sur les ordinateurs actuels.
La méthode de calcul proposée ci-dessous est dite de
Monte-Carlo. Pour qui ne serait pas familier de cette tech-
nique, on signalera qu’il s’agit d’une méthode de simula-
tion basée sur un générateur de nombres aléatoires comme
on en trouve dans la plupart des langages de programma-
tion. Un appel ou une série d’appels à une telle fonction
random permet de reproduire un événement du phéno-
mène étudié selon la loi de probabilité qui est la sienne
(nous indiquerons dans le texte comment cela peut se faire
dans le cas qui nous occupe). Sachant comment reproduire
un événement, on en génère un nombre plus ou moins
élevé, de façon à reconstruire peu à peu l’ensemble de la
distribution. Les procédures Monte-Carlo conviennent
particulièrement aux distributions non analytiques, c’est-
à-dire auxquelles on ne peut appliquer facilement les
outils mathématiques standards de dérivation, d’intégra-
tion... Toutefois, contrairement aux calculs analytiques qui
peuvent toujours être considérés comme très précis, sinon
exacts, l’usage de nombres aléatoires conduit à des résul-
tats fluctuants dont la précision dépendra directement du
nombre d’événements générés : plus nombreux ils sont,
plus les fluctuations relatives seront réduites. On pourrait
en déduire que l’usage d’un Monte-Carlo suppose de
longs temps de calcul, mais il faut souligner d’une part,
que bon nombre d’applications ne demandent pas après
tout de précision très élevée et, d’autre part, que les ordi-
nateurs actuels possèdent une puissance de calcul remar-
quable. On notera également que la question se pose diffé-
remment selon qu’on ait affaire à une opération qui doit se
faire une fois pour toutes, et à laquelle on pourra donc
consacrer un très long temps de calcul, ou à une opération
fréquemment utilisée en routine et qui se doit donc d’être
relativement courte.
L’approche usuelle
Nous reprenons ici un point développé dans la référence 1,
en l’adaptant à la présente discussion.
Supposons une bactérie inconnue qui donne pour les n
tests un résultat R(n) = {R
1
,R
2
, ...R
j
, ...R
n
}. Dans cette
notation, R
j
est la réponse, positive ou négative, de la
bactérie au test numéro j. Soit B
ij
la probabilité pour la
bactérie numéro i dans le tableau d’identification de don-
ner le même résultat que celui qui est observé.
(1)
Bij =Aij
(
si Rjpositif
)
Bij =1−Aij
(
si Rjnégatif
)
Selon la procédure usuelle [8, 9], les B
ij
sont alors multi-
pliés entre eux pour donner :
(2)
Li=Bi1Bi2...Bin =
写
j=1
n
Bij
Dans l’hypothèse où les tests sont tous indépendants, ces
quantités ont une signification précise, dépourvue d’ambi-
guïté. Chaque L
i
représente la probabilité pour la bactérie
i de fournir le profil observé. À l’évidence, une probabilité
élevée devrait conduire à retenir la candidate, tandis
qu’une probabilité faible devrait suggérer de la rejeter.
Mais, sachant que nous avons presque toujours affaire à de
petits nombres la question devient : Quelle probabilité doit
être considérée comme élevée, et laquelle doit être vue
comme faible ? Ici intervient l’un des aspects les plus
discutables de la méthode classique, qui propose de com-
parer entre elles les valeurs L
i
, de sorte que le taxon qui
obtient un résultat élevé sera considéré comme un
meilleur candidat que celui qui obtient un résultat faible.
Mais le fait est que les L
i
ne sont que grossièrement com-
parables. Certaines bactéries présentent une grande varia-
bilité aux tests, ce qui se marque par un faible pouvoir
discriminant des nombres A
ij
(par exemple 51 %-49 %, ou
75 %-25 %), et en définitive par de faibles probabilités
pour tous les profils possibles, y compris le plus favorable.
Par contre, d’autres bactéries peuvent offrir des profils de
meilleure qualité aux probabilités élevées. Ainsi peut-il se
faire que deux taxons obtiennent des résultats compara-
bles, ce résultat étant le plus haut possible pour l’un mais
se révélant peu convaincant pour l’autre. Or, dans la com-
paraison qu’on nous propose, ces deux candidats seront
mis sur un pied d’égalité. Nous trouvons ici la raison pour
laquelle la discussion est souvent complétée par une infor-
mation auxiliaire telle que le biotype, qui permet d’appré-
cier la qualité d’un résultat en comparant celui-ci au
meilleur profil de chaque candidat. Indépendamment de
son efficacité, l’ensemble de la procédure paraît peu élé-
gante et la raison en est l’absence de valeurs de référence
capables de conférer à la décision finale un niveau de
confiance clairement défini.
Un autre point qui peut être discuté est l’usage selon
lequel les valeurs (2) sont souvent présentées sous une
forme normalisée, à savoir :
pratique quotidienne
Ann Biol Clin, vol. 64, n° 1, janvier-février 200684
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

(3)
Mi=Li
L1+L2+... +Ln
=Li
兺
k=1
n
Lk
avec M1+M2+... +Mn=1
Puisque l’idée est de comparer entre elles les bactéries, les
M
i
sont effectivement plus commodes à discuter que les
L
i
, lesquels sont souvent de très petits nombres. Les M
i
sont souvent appelés scores d’identification des différents
candidats. Il est important de souligner que ces scores ne
sont rien d’autre que le résultat d’une procédure de norma-
lisation et qu’ils ne peuvent en aucune façon apporter plus
d’information que les L
i
. Au contraire, ils perdent dans
l’opération le statut de valeurs de probabilité. Le problème
ici est que, précisément, ils ont toute l’apparence de
valeurs de probabilité et il est vraisemblable qu’ils sont
habituellement vus et appréciés comme tels. Pour illustrer
ce point, supposons un cas d’identification qui propose
deux candidats A et B avec M
A
=97% et M
B
=3%.
L’argument qui tend à prévaloir est que si nous retenons A
chaque fois que se présente le profil observé, alors nous
nous tromperons seulement trois fois parmi cent. Or ceci
n’est pas correct. À supposer par exemple que A soit une
bactérie beaucoup plus rare que B, alors le risque d’erreur
peut être beaucoup plus élevé que 3 %, point important
qui sera développé en annexe de cet article (Annexe 1).On
voit qu’ici le défaut est un peu l’inverse de celui du para-
graphe précédent : les chiffres présentés semblent pouvoir
être associés facilement à un risque d’erreur, ce qui n’est
pas le cas.
Puisque la comparaison entre bactéries n’est que pauvre-
ment justifiée, il apparaît qu’une meilleure voie serait de
discuter chacune d’elles indépendamment des autres. Plu-
tôt que de poser la question : « Quelle est la meilleure
candidate ? », nous devrions nous demander pour chaque
bactérie s’il s’agit, oui ou non, d’une candidate plausible.
Ceci revient à apprécier si le profil observé, mesuré par sa
probabilité L
i
, est suffisamment typique de la i
e
bactérie,
ou si au contraire il lui est plutôt atypique. Nous sommes
donc amenés à étudier la distribution de probabilité des L
i
pour chaque bactérie en particulier et à définir sur cette
distribution une valeur seuil qui permette de l’accepter ou
de la rejeter.
La distribution de probabilité
des valeurs L
i
Nous nous intéressons à la ligne du tableau d’identifica-
tion qui concerne le seul taxon numéro i, de sorte que dans
la suite nous omettrons l’indice i dans les notations (A
ij
devient A
j
,L
i
devient L, etc.). Pour chaque test, le micro-
organisme concerné peut répondre soit positivement soit
négativement, ce qui pour n tests conduit à 2
n
résultats
possibles pour la formule (2). Ainsi une galerie de 30 tests
pourrait mener à plus d’un milliard de valeurs, la plupart
d’entre elles étant en fait très proches de zéro. Notons que
nous pouvons les classer de la manière suivante : soit D
j
la
réponse la plus probable au test j, et soit d
j
la réponse la
plus basse à ce même test :
(4)
Dj=Aj
(
si Aj≥0,5
)
Dj=1−Aj
(
si Aj<0,5
)
dj=1−Dj
À présent, pour classer les valeurs L nous commençons
par la plus probable, en soulignant que “ la plus probable ”
est souvent loin de signifier “ hautement probable ” !
(5)
Lmax =D1D2......Dn=
写
j=1
n
Dj
Cette valeur unique peut être dite d’ordre zéro. La classe
suivante est faite des n valeurs d’ordre 1, à savoir les
produits où la réponse à un et un seul test n’est pas la plus
probable :
(6)
dk
写
j=1
j≠k
n
Dj
Le second ordre sera fait bien entendu des n(n-1) produits
où deux et seulement deux réponses ne sont pas les plus
probables :
(7)
dkd1
写
j=1
j≠k,1
n
Dj
La manière de poursuivre cette classification est évidente.
Pour en venir à la représentation graphique de l’ensemble
de la distribution, on notera que sa principale caractéristi-
que est que chaque valeur L représente sa propre probabi-
lité d’apparaître. La conséquence de ceci est que l’allure
générale du graphe suivra la droite de pente 1. Les excep-
tions à cette tendance viendront de profils différents dont
les valeurs sont égales ou très voisines et donc se superpo-
seront. Ceci devrait être assez fréquent dans la partie basse
de la distribution, où de nombreuses valeurs faibles sont
très proches les unes des autres.
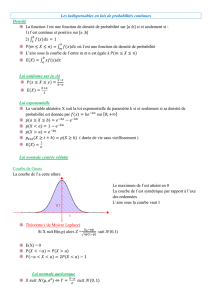
La figure 1 montre un exemple de distribution obtenue
avec un ensemble très limité de 10 tests. Peu importe le
micro-organisme concerné, les nombres Aj utilisés, expri-
més en pourcentage, étaient : 99, 50, 68, 99, 43, 86, 4, 40,
99, 1.
Risque d’erreur en identification bactérienne
Ann Biol Clin, vol. 64, n° 1, janvier-février 2006 85
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

Deux points sont remarquables sur la distribution : la
limite haute, donnée par l’équation (5), et la valeur
moyenne donnée par :
(8)
L
¯
=
写
j=1
n
[
Aj
2+
(
1−Aj
)
2
]
Pour décider si un profil observé peut être raisonnable-
ment considéré comme typique de la distribution, et donc
si la bactérie est un candidat plausible à l’identification,
nous sommes amenés à définir un niveau seuil tel que
toute valeur supérieure à ce niveau seuil serait acceptée,
alors qu’elle serait rejetée si elle se révèle inférieure. Ceci
répond à la notion très classique de risque d’erreur alpha,
ou de première espèce, qui mesure la probabilité de rejeter
la solution correcte à un problème [10]. Un risque alpha
de 5 % par exemple suppose que la distribution présente
5 % de sa surface sous le niveau choisi et 95 % au-delà, de
sorte que parmi toutes les occurrences de la bactérie
concernée, nous la rejetterons comme candidate valable et
nous tromperons ainsi cinq fois sur cent.
Nous proposons ci-dessous d’utiliser le calcul Monte-
Carlo pour calculer l’ensemble de la distribution de proba-
bilité et y définir le seuil de référence.
Calcul Monte-Carlo
Dans un calcul Monte-Carlo, des nombres aléatoires sont
générés pour simuler les réponses aux différents tests,
obéissant aux probabilités A
j
de façon à fournir un profil
plausible pour la bactérie concernée. Le programme que
nous avons utilisé a été rédigé en langage VBA (visual
basic for applications) dans l’environnement du tableur
Microsoft Excel
®
. Dans ce langage, le générateur de nom-
bres aléatoires est la fonction rnd(). Chaque appel du
genre N = rnd() à cette fonction fournit un nombre N com-
pris entre 0 et 1. Si N est plus petit que ou égal à A
j
,on
convient qu’on a obtenu une réponse positive au j
e
test ;
s’il est plus grand, la réponse au test est considérée
comme négative. On voit que n appels à rnd() permettent
de simuler les réponses à l’ensemble des n tests et donc
d’obtenir un profil plausible de la bactérie.
En répétant un grand nombre de fois la procédure, on
reproduit les différents profils, chacun d’eux apparaissant
à la fréquence qui lui est propre. Le spectre complet des
réponses peut ainsi être construit, avec une précision qui
dépendra du nombre d’événements simulés. C’est de cette
manière que la figure 1 a été obtenue, sur base de 10
5
événements.
Connaissant en détails l’ensemble de la distribution, il est
facile de compléter le programme en lui demandant d’y
localiser une valeur qui réponde à un critère donné. Ainsi,
si nous demandons un niveau de risque alpha égal à 1 %
(le niveau choisi est à vrai dire purement conventionnel), il
s’agit de trouver la valeur qui partage en deux la surface
de la distribution, la partie basse représentant un pour cent
du total, ou en tout cas la portion la plus proche possible
de ce résultat, compte tenu de la résolution atteinte dans le
calcul. Cette valeur servira de niveau de référence pour
décider des candidats valables et des candidats à rejeter.
0
10 000
20 000
30 000
40 000
0 0,05 0,1 0,15 0,2 0,25 0,3
dN/dL
Distribution de 105 événements
générés aléatoirement
0
50
100
150
200
250
0,0005 0,001
Valeurs des Li
Valeurs des Li
dN/dL Ra (1%) = 4,7x10-4
Lmax = 0,31
Figure 1. Distribution de 10
5
événements pour dix tests dont les
A
i
seraient (en %) : 99, 50, 68, 99, 43, 86, 4, 40, 99, et 1. Le
graphe inférieur est un zoom sur la partie basse de la distribu-
tion.Onyaindiquéleniveauderisque alpha (R
a
) correspondant
à 1 % d’erreur.
pratique quotidienne
Ann Biol Clin, vol. 64, n° 1, janvier-février 200686
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

Il n’est pas nécessaire d’atteindre une grande précision
dans la détermination du niveau de risque, compte tenu du
caractère relativement arbitraire de ce choix. Il faut souli-
gner toutefois que la puissance de calcul devenue très
commune sur les ordinateurs actuels permettrait très cer-
tainement d’être exigeant sur la précision finale : sur
microprocesseur cadencé à 3,2 GHz, la routine utilisée
pour construire la figure 1 prend 87,5 secondes par bacté-
rie pour le calcul de 10
5
événements sur une galerie de 33
tests. En soulignant que l’opération ne doit être menée
qu’une fois pour toutes sur une galerie donnée, le temps
de calcul apparaît parfaitement raisonnable, même pour
un nombre élevé de bactéries.
Procédure d’aide à l’identification
L’approche théorique qui précède suggère différentes pro-
cédures susceptibles d’apporter en pratique une aide
mathématique à l’identification bactérienne. La première
serait facile à mettre en œuvre quelles que soient les possi-
bilités de calcul disponibles en routine : partant du tableau
d’identification de la galerie concernée, la distribution des
probabilités L
i
de chaque bactérie serait générée par calcul
Monte-Carlo. Sur ces distributions seraient repérées une
fois pour toutes les niveaux de risque alpha correspondant
à un critère prédéfini (par exemple : 5 % d’erreur. Rien
n’empêche de définir plusieurs niveaux de risque de façon
à laisser au praticien une certaine liberté de décision).
Lors du travail en routine, les réponses observées seraient
confrontées aux niveaux calculés : les bactéries dont les
probabilités L
i
apparaîtraient supérieures à ces niveaux
seraient considérées comme des candidates valables à
l’identification. Les autres seraient rejetées.
Si plusieurs résultats étaient à retenir comme plausibles, il
y aurait lieu d’appliquer les méthodes de discrimination
habituelles (tests complémentaires...)
Une autre procédure plus satisfaisante encore suppose que
la puissance de calcul soit disponible en routine : pour
toute identification, il s’agirait de situer la réponse du
germe inconnu dans les différentes distributions de proba-
bilité des germes candidats et de calculer la probabilité
qu’il fournisse un résultat plus petit ou égal à ce qu’on
observe. On aurait ici la possibilité de remplacer le tradi-
tionnel ensemble de valeurs L
i
ou M
i
, dont on a souligné
les défauts, par un ensemble de nombres dont la significa-
tion statistique serait claire : voir un germe candidat asso-
cié au nombre 0,5 par exemple signifierait que, s’il s’agit
du germe inconnu, il ne pourrait donner un meilleur score
qu’une fois sur deux, de sorte qu’il n’y aurait pas là
d’argument raisonnable pour le rejeter. Le nombre 0,005
signifierait que, s’il s’agit du germe inconnu, il a fourni un
résultat très peu probable ; plus vraisemblablement, la
vérité est ailleurs. Un nombre tel que 0,05 serait laissé à
l’appréciation du praticien, puisque la probabilité de faire
moins bien est faible... mais correspond tout de même à
une occurrence sur 20.
Somme toute, si la première procédure se base sur des
niveaux de référence associés à chaque bactérie, la
seconde permet de définir un véritable niveau de confiance
à associer à la décision finale. Elle suppose, comme on l’a
dit, que les spectres de toutes les bactéries soient explorés
lors de chaque identification. Première possibilité : un cal-
cul Monte-Carlo est généré sur chaque profil mesuré en
routine. L’estimation du paragraphe précédent montre
qu’il est possible d’obtenir en un temps raisonnable, à
savoir une seconde environ par bactérie, la simulation de
1 000 événements représentatifs de cette bactérie, ce qui
devrait suffire à lui attribuer le niveau de confiance requis.
Autre possibilité : les distributions de probabilité sont cal-
culées une fois pour toutes et stockées en mémoire. Lors
d’une identification en routine, chaque valeur L
i
est locali-
sée sur la distribution correspondante. Le niveau de
confiance est alors obtenu par la somme des valeurs de la
distribution inférieures ou égales à L
i
.
Conclusion
Il est possible d’apporter une correction à la méthode
usuelle d’aide mathématique à l’identification bactérienne
grâce à la construction des spectres individuels des diffé-
rents germes, construction qui peut se faire par calcul
Monte-Carlo. La décision de retenir ou de rejeter une
bactérie se base alors uniquement sur des caractéristiques
qui lui sont propres et évite la comparaison avec les autres,
comparaison souvent peu justifiée. L’intégrale de la partie
basse de chaque spectre fournit un nombre dont l’interpré-
tation est directe : dans tous les cas où la bactérie concer-
née se présenterait effectivement à l’identification, quel
serait notre taux d’erreur si nous rejetions tous les nom-
bres inférieurs au résultat calculé ? En pratique, différen-
tes stratégies sont envisageables : ou bien on calcule une
fois pour toutes des niveaux de référence à ne pas dépas-
ser, ou bien on évalue lors de chaque mesure de routine de
véritables niveaux de confiance liés aux différents germes,
niveaux de confiance qui pourraient remplacer avantageu-
sement les jeux de valeurs (2) ou (3). Cette dernière sug-
gestion suppose une puissance de calcul que nous croyons
à la portée des micro-ordinateurs actuels.
Remerciements.Je remercie mes collègues Corinne
Meidner et Maurice Meunier pour leur aide précieuse et
avis éclairé.
Risque d’erreur en identification bactérienne
Ann Biol Clin, vol. 64, n° 1, janvier-février 2006 87
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.
 6
7
6
7
1
/
7
100%