Une hypertension artérielle pulmonaire révélatrice d`une

Arrêt sur image
Sang Thrombose Vaisseaux 2009 ;
21, n° 2 : 99-101
doi: 10.1684/stv.2009.0366
Une hypertension arte
´rielle pulmonaire
re
´ve
´latrice d’une hyperthyroı
¨die
Hyperthyroidism presenting with pulmonary hypertension
El Mehdi Badidi
Service de cardiologie du 5
e
hôpital militaire, Fès, Maroc
Observation
Une patiente âgée de 46 ans, sans antécédents pathologiques notables s’était
présentée, il y a un an, dans un tableau de dyspnée classe III de la New York
Heart Association (NYHA) et d’insuffisance cardiaque droite d’installation
rapidement progressive dans un contexte d’altération de l’état général et
d’amaigrissement important. Le diagnostic d’hypertension pulmonaire
(HTAP) compliquant une hyperthyroïdie était retenu sur les données biolo-
giques par le profil hormonal : TSH effondrée à 0,005 UI, T4 à 120,3 UI. Un
traitement associant Néo-Mercazole
®
, bêtabloquants, antivitamines K, anti-
aldostérone et diurétique de l’anse était instauré.
Après un an de traitement, la patiente rapportait une légère amélioration clinique.
Al’examen, on notait une régression des signes de congestion droite. L’électrocar-
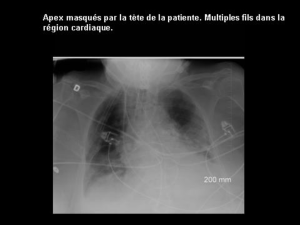
diogramme inscrivait une AC/FA avec réponse ventriculaire à 75 cpm. Le cliché
thoracique montrait la persistance de la cardiomégalie (RCT = 0,64) (figure 1).
L’échocardiographie mode 2D objectivait une dilatation importante des cavités
Tire
´sa
`part :
EM Badidi Figure 1.A) Scanner : dilatation aux dépens des cavités droites ; B) radiographie mon-
trant une cardiomégalie.
99
STV, vol. 21, n°2, fe
´vrier 2009
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

droites : oreillette droite ectasique (surface OD = 40 cm
2
),
ventricule droit, artère pulmonaire et ses deux branches
(figure 2) avec un épanchement péricardique de faible abon-
dance. Au Doppler, une HTAP importante à 80 mmHg de
pression artérielle pulmonaire était mise en évidence (PAP).
Les cavités cardiaques gauches étaient refoulées.
Figure 2.A) Coupes 4 cavités objectivant une ectasie de l’OD ; B) coupe parasternale G montrant une dilatation importante de l’artère
pulmonaire.
Figure 3.Scanner objectivant la dilatation des deux branches de l’artère pulmonaire.
100 STV, vol. 21, n°2, fe
´vrier 2009
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.

L’angioscanner thoracique objectivait une dilation importante
de l’artère pulmonaire (AP) et de ses branches ainsi que de la
veine cave inférieure (figures 1, 3).
Commentaires
Le mécanisme étiopathogénique de l’HTAP au cours de
l’hyperthyroïdie demeure imprécis [1]. L’hypothèse la
plus séduisante implique l’action d’hormones thyroïdien-
nes via une potentialisation de l’action des catécholamines
et des substances vasodilatatrices (NO et prostacycline) au
niveau de l’arbre artériel pulmonaire [1, 2].
Sur le plan clinique, la dyspnée peut être le témoin de
l’HTAP et de son évolution, ou représenter une manifesta-
tion de l’hyperthyroïdie. Le traitement repose sur les bêta-
bloquants et le traitement spécifique de l’hyperthyroïdie [2,
3]. Les antithyroïdiens de synthèse, en particulier le méthi-
mazole (métabolite du carbimazole), représenteraient le
traitement de choix, car il s’agit du moyen le plus rapide
de normaliser la pression artérielle pulmonaire, en compa-
raison à l’iode radioactif ou à la chirurgie. L’évolution habi-
tuellement favorable avec le retour à un état euthyroïdien ne
se produit pas chez notre patiente [3]. ■
Références
1. Yanai-Landau H, Amital H, Bar-Dayan Y, et al. Autoimmune aspects
of primary pulmonary hypertension. Pathophysiology 1995 ; 63 : 71-5.
2. Fischer J. Hypothyroidism and primary pulmonary hypertension. Ann
Intern Med 1994 ; 120 : 167-8.
3. Thurnheer R, Jenni R, Russi EW, Greminger P, Speich R. Hyperthyroi-
dism and pulmonary hypertension. J Intern Med 1997 ; 242 : 185-8.
101
STV, vol. 21, n°2, fe
´vrier 2009
Copyright © 2017 John Libbey Eurotext. Téléchargé par un robot venant de 88.99.165.207 le 24/05/2017.
1
/
3
100%