Comment dépister une cardiopathie congénitale à l`âge

28 | La Lettre du Cardiologue • n° 454-455 - avril-mai 2012
DOSSIER THÉMATIQUE
Prise en charge
descardiopathies congénitales
de l’enfant devenu adulte
* Cardiologie pédiatrique et congéni-
tale adulte, unité d’exploration cardio-
vasculaire de l’Orangerie, Strasbourg.
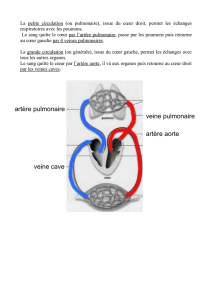
Comment dépister
une cardiopathie congénitale
à l’âge adulte ?
Diagnosis of native congenital heart disease in the adult
J. Radojevic*
L
es progrès de la cardiopédiatrie et de la chirurgie
cardiaque ont permis, depuis un demi-siècle,
que plus de 85 % des enfants porteurs de
cardiopathies congénitales (CC) atteignent l’âge
adulte (1). La population des patients adultes
porteurs de CC est en constante progression : elle
est actuellement estimée à plus de 1,2 million en
Europe (2).
Même si la plupart des patients diagnostiqués
dans l’enfance nécessitent un suivi à vie (3), nous
nous intéresserons dans cet article aux cas moins
fréquents des cardiopathies qui ne sont diagnosti-
quées qu’à l’âge adulte (4). L’objectif est de familia-
riser le lecteur avec leurs modes de présentation et
leurs prises en charge qui peuvent être complexes
et sont souvent faites sur mesure. Pour une prise en
charge optimale, la coordination entre le cardiologue
et le centre spécialisé dans les cardiopathies congé-
nitales de l’adulte est essentielle. Un schéma de suivi
en fonction de la complexité de la cardiopathie a été
proposé (tableau) [5].
Il ne s’agit pas d’une revue exhaustive. Les recom-
mandations thérapeutiques citées dans cet article
sont fondées, pour la plupart, sur les études pros-
pectives non randomisées, les études rétrospectives
et les opinions d’experts.
Tableau. Suivi d’une cardiopathie congénitale à l’âge adulte en fonction de sa complexité (adapté à partir de Warnes CA4).
Cardiopathies congénitales simples :
suivi dans un centre spécialisé non nécessaire
Cardiopathies congénitales de complexité modérée :
suivi alterné avec un centre spécialisé nécessaire
Cardiopathies congénitales complexes :
suivi exclusif dans un centre spécialisé
Natives
Valvulopathie aortique isolée
Valvulopathie mitrale isolée (excepté valve mitrale
parachute et fente mitrale)
Petite communication CIA
Petite communication CIV isolée
Sténose pulmonaire légère
Traitées
Ligature (occlusion) du canal artériel persistant
CIA ostium secundum ou sinus venosus réparée
sans séquelles
CIV réparée sans séquelles
Fistules entre l’aorte et le ventricule gauche
Retour veineux pulmonaire anormal (partiel ou total)
Canal AV (partiel ou complet)
Coarctation de l’aorte
Maladie d’Ebstein
Sténose infundibulaire significative
Canal artériel persistant (non fermé)
Insuffisance pulmonaire (modérée et sévère)
Sténose pulmonaire (modérée et sévère)
Anévrisme/fistule du sinus de Valsalva
CIA type sinus venosus
Sténose sous- et supravalvulaire aortique (sauf CMO)
Tétralogie de Fallot
CIV avec : atrésie des valves, insuffisance aortique, coarctation
de l’aorte, maladie mitrale, sténose infundibulaire,
straddling
desvalves AV, sténose sous-aortique
Présence de tubes valvés ou non valvés
Cardiopathies cyanogènes (toutes)
Ventricule droit à double issue
Syndrome d’Eisenmenger
Circulation de type Fontan
Atrésie mitrale
Ventricule unique
Atrésie pulmonaire
Transposition des gros vaisseaux
Atrésie tricuspide
Truncus arteriosus
Autres anomalies de connexion ou de situs :
cœur
criss-cross
, isomérisme et hétérotaxie,
inversion ventriculaire
AV : atrioventriculaire; CIA : communication interauriculaire; CIV : communication interventriculaire; CMO : cardiomyopathie obstructive.

Figure 2. Communication interauriculaire sinus venosus.
Figure 1. Dilatation des cavités droites et communication interauriculaire ostium secondum.
OD : oreillette droite ; OG : oreillette gauche ; VCS : veine cave supérieure.
La Lettre du Cardiologue • n° 454-455 - avril-mai 2012 | 29
Résumé
Summary
About ten percent of congenital
heart diseases will be discov-
ered only in adulthood. In
this article, we focus on their
diagnostic workout and their
therapeutic options. The aim is
to enable readers to recognize
them, and to provide the best
therapeutic options. The recom-
mendations in this article are
based mostly on the results of
non-randomized prospective
and retrospective studies and
on expert opinion.
Keywords
Native congenital heart
disease
Adult
Diagnosis
Mots-clés
Cardiopathie
congénitale native
Adulte
Dépistage
Même si la plupart des cardiopathies congénitales sont diagnostiquées dans l’enfance, environ 10 % d’entre
elles ne seront découvertes qu’à l’âge adulte de façon fortuite, ou lors des suites d’une complication
évolutive. L’objectif de cet article est de familiariser le lecteur avec le mode de présentation clinique, le
diagnostic échocardiographique et la prise en charge optimale de ces cardiopathies restées “silencieuses”.
Les recommandations utilisées dans le texte sont fondées sur les études prospectives non randomisées,
les études rétrospectives et les opinions d’experts.
Dilatation des cavités droites :
communication interauriculaire
et/ou retour veineux
pulmonaire anormal
La communication interauriculaire (CIA) peut ne pas
être diagnostiquée jusqu’à l’âge adulte en raison de
sa bonne tolérance et de l’absence de symptômes. La
plupart des patients développent des complications
(dyspnée d’effort, arythmie supraventriculaire et
insuffisance cardiaque [IC] droite) après 40 ans (6).
Cliniquement, on observe un souffle systolique au
foyer pulmonaire, associé à un dédoublement du B2.
À l’échographie cardiaque, le signe d’appel est
la présence d’une dilatation des cavités droites
(figure 1). Si une CIA de type ostium secundum
(60 %) ou ostium primum (20 %) n’est pas visible,
il faut penser à la coupe sous-costale bicave, afin
de rechercher une CIA de type sinus venosus (15 %)
avec un retour veineux pulmonaire anormal inva-
riablement associé (figure 2). Cette vue peut être
difficile à obtenir chez un patient adulte ; dans ce cas,
une échographie transœsophagienne avec incidence
bicave à 90° complétera utilement le bilan. Devant
une dilatation des cavités cardiaques droites, mais
sans CIA, il faut penser à un retour veineux pulmo-
naire anormal partiel isolé, et confirmer le diagnostic
par un angioscanner thoracique (figure 3, p. 30).
Quel que soit l’âge du patient, la découverte d’une
CIA avec une dilatation des cavités droites et un
shunt gauche-droit, en l’absence d’une hyperten-

Figure 4. Coarctation de l’aorte (mode 2D, doppler couleur et doppler continu).
Figure 3.
Communication
interauriculaire sinus
venosus. Retour veineux
pulmonaire anormal
partiel dans la veine
cave supérieure (vue au
scanner).
Ao : aorte ; AP : artère pulmonaire ; OG : oreillette gauche ;
VCS : veine cave supérieure ; VPMD : veine pulmonaire moyenne ;
VPSD : veine pulmonaire supérieure droite.
30 | La Lettre du Cardiologue • n° 454-455 - avril-mai 2012
Comment dépister unecardiopathie congénitale àl’âge adulte ?
DOSSIER THÉMATIQUE
Prise en charge
descardiopathies congénitales
de l’enfant devenu adulte
sion artérielle pulmonaire (HTAP) fixée, doit faire
poser l’indication de la fermeture (6). Pour les
CIA de type ostium secundum, une fermeture par
cathétérisme interventionnel est actuellement la
méthode de choix, avec prévention antioslérienne
et traitement par aspirine à dose antiagrégante
pendant 6 mois après la fermeture (3). Pour tous
les autres types de CIA, la fermeture sera chirur-
gicale. Il faut se méfier d’une dysfonction ventri-
culaire gauche (VG), surtout chez le sujet âgé, car
la fermeture de la CIA peut précipiter une poussée
d’insuffisance VG.
Les bénéfices attendus de la fermeture des CIA
à l’âge adulte sont la réduction de nombreuses
décompensations cardiaques et l’amélioration
des capacités à l’effort et de la qualité de vie. Le
risque de survenue d’arythmies supraventriculaires
persiste après la fermeture, et d’autant plus que
la fermeture est tardive (après l’âge de 25 ans),
mais leur tolérance est meilleure, et leur gestion
plus facile (7).
S’il y a une indication à l’ablation d’une fibrillation
auriculaire, il vaut mieux la réaliser avant la ferme-
ture de la CIA, en raison de la nécessité d’un abord
transseptal.
Hypertension artérielle
chez le sujet jeune :
coarctation de l’aorte
Les patients adultes porteurs d’une coarctation de
l’aorte (CoA) sont le plus souvent asymptomatiques.
Les éventuels symptômes traduisent une hyperten-
sion de la partie supérieure et une hypoperfusion de
la partie inférieure du corps (céphalées, épistaxis,
acouphènes, vertiges, pieds froids, douleurs abdo-
minales, faiblesse des membres inférieurs à l’effort).
Le diagnostic d’une CoA est clinique et repose sur
l’existence d’un gradient de pression de plus de
20 mmHg entre les membres supérieurs et infé-
rieurs (3). D’autres signes cliniques sont à rechercher :
un délai entre le pouls radial et le pouls fémoral, un
pouls fémoral faible, les collatérales palpables sur la
paroi thoracique, un souffle continu interscapulaire
gauche et un frémissement suprasternal.
En échographie cardiaque, on visualise, en vue supra-
sternale, un rétrécissement de l’isthme aortique avec
un flux aliasé en doppler couleur. Le signe pathogno-
monique de la CoA est la présence d’un prolongement
diastolique en mode doppler continu (figure 4) [3].

Figure 5. Membrane sous-valvulaire aortique.
Figure 6. Sténose supra-
valvulaire aortique,
syndrome de Williams
et Beuren.
La Lettre du Cardiologue • n° 454-455 - avril-mai 2012 | 31
DOSSIER THÉMATIQUE
Chez un sujet jeune hypertendu souffrant de claudi-
cation des membres inférieurs, il faut chercher un
CoA de l’aorte abdominale.
Les anomalies associées à rechercher sont : les
obstacles étagés du cœur gauche, la bicuspidie
aortique (jusqu’à 80 % des CoA), la dilatation de
l’aorte ascendante, l’hypertrophie ventriculaire
gauche, la communication interventriculaire.
Le traitement d’une CoA native chez l’adulte est
encore débattu, mais, le plus souvent, il est endo-
vasculaire par angioplastie et mise en place d’un
stent. Le traitement par aspirine à dose antiagré-
gante, la prévention antioslérienne et la surveil-
lance tensionnelle sont indiqués à vie (un tiers
des patients restent hypertendus, malgré la levée
optimale de la CoA [8]).
Souffle systolique éjectionnel
avec valve aortique normale :
sténose sous-valvulaire
et supravalvulaire aortique,
sténose pulmonaire
La sténose sous-valvulaire aortique représente
jusqu’à 30 % des obstacles de la voie d’éjection
gauche (9). C’est une lésion d’évolution progressive,
qui peut ne pas être découverte jusqu’à l’âge adulte.
La lésion causale est une membrane fibreuse loca-
lisée (figure 5) ou, plus rarement, un tunnel fibro-
musculaire. Une anomalie de l’insertion de l’appareil
sous-valvulaire mitral peut participer à l’obstruction.
Un bilan lésionnel précis est important et déter-
mine la stratégie chirurgicale. Il ne faut pas oublier
d’examiner toute la voie gauche, car l’obstacle du
cœur gauche peut être étagé (supra- et sous-valvu-
laire mitral, supra- et sous-valvulaire aortique, et
coarctation de l’aorte). Le risque à long terme est
la récidive (jusqu’à 30 % des cas), et une progres-
sion vers l’insuffisance et vers la sténose valvulaire
aortique (9-11). La sténose supravalvulaire aortique
est plus rare (8 % des obstacles d’éjection gauche).
Elle est le plus souvent associée à un syndrome de
Williams et Beuren (9). L’aorte a typiquement un
aspect en sablier, la sténose se situant au niveau
de la jonction sinotubulaire (figure 6).
Un gradient doppler moyen supérieur à 50 mmHg
chez un patient symptomatique, un patient asymp-
tomatique mais avec une hypertrophie ventriculaire
gauche, une fraction d’éjection inférieure à 50 %, une
insuffisance aortique sévère avec dilatation VG ou
une réponse tensionnelle anormale à l’effort sont des
indications à la chirurgie pour les sténoses sous- et
supravalvulaires aortiques (3).
La sténose valvulaire pulmonaire représente 10 %
des cardiopathies congénitales, avec 2 à 3 % de
récurrences familiales (3). L’aspect typique est celui
de la valve en dôme (figure 7, p. 32). Une dilata-
tion du tronc et de l’artère pulmonaire (AP) gauche
peuvent être associées. Plus rarement, la valve est
dysplasique et épaissie, et peut s’associer à une
sténose supravalvulaire en sablier, comme dans
le syndrome de Noonan. Une vitesse supérieure à
4 m/s (gradient maximum supérieur à 64 mmHg)
avec un ventricule droit (VD) normofonctionnel
est habituellement retenue comme indication
de l’intervention (3). Pour une sténose valvulaire
typique, une dilatation percutanée au ballon est la
procédure de choix.
Un autre obstacle de la voie d’éjection du VD est
la sténose médioventriculaire (double chamber

Figure 8. Canal artériel persistant en doppler couleur (A) et doppler continu (B).
A B
Figure 7. Sténose valvaire pulmonaire. Valve épaisse, gradient maximum élevé.
32 | La Lettre du Cardiologue • n° 454-455 - avril-mai 2012
Comment dépister unecardiopathie congénitale àl’âge adulte ?
DOSSIER THÉMATIQUE
Prise en charge
descardiopathies congénitales
de l’enfant devenu adulte
right ventricle). Cette anomalie rare est souvent
associée à une communication interventriculaire
périmembraneuse. L’obstacle est créé par l’hyper-
trophie musculaire sous-infundibulaire, qui divise le
VD en une chambre à haute pression en amont de
l’obstacle et une chambre à basse pression en aval.
Lorsqu’elle est présente, l’insuffisance tricuspide
est véloce et peut, à tort, faire poser le diagnostic
d’HTAP. La direction du shunt de la communication
interventriculaire est déterminée par le rapport des
pressions VG et VD en amont de l’obstacle. Une
vitesse supérieure à 4 m/s à travers l’obstacle est
une indication opératoire (3).
Souffle continu
avec une échographie cardiaque
“normale” : canal artériel
persistant ou fistule coronaire
Le canal artériel persistant (CAP) est une communi-
cation entre l’AP gauche proximale et l’aorte descen-
dante en aval de l’artère sous-clavière gauche. Les
CAP de grande et de moyenne taille se compliquent
d’une IC gauche et d’une HTAP (syndrome d’Eisen-
menger). Les CAP de petite taille (dits restrictifs)
peuvent être asymptomatiques, avec à l’examen
clinique un souffle continu dans la région sous-
clavière gauche. Le risque évolutif principal est alors
une endocardite infectieuse (12, 13).
L’échographie cardiaque confirme le diagnostic en
montrant un flux rétrograde dans l’artère pulmo-
naire en vue parasternale gauche petit axe, et un
vaisseau sous la crosse aortique (figure 8A) avec flux
continu (figure 8B). La taille et la fonction du VG,
ainsi que la pression artérielle pulmonaire, doivent
être évaluées.
Un canal artériel “soufflant” est une indication à la
fermeture percutanée quelle que soit sa taille (la
chirurgie du canal est très risquée chez l’adulte).
En l’absence de dilatation ou de dysfonction ventri-
culaire gauche et d’HTAP, aucun suivi au-delà de
6 mois après la fermeture n’est nécessaire (3).
Les fistules coronaires sont des communications
entre le réseau des artères coronaires et les cavités
 6
7
8
6
7
8
1
/
8
100%