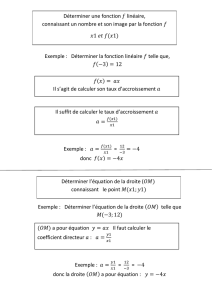
Chimie Physique

Une nouvelle édition rénovée et accomplie de
l’ouvrage de référence de la chimie physique
Cette 4eédition du traité de Chimie physique (Atkins’ physical
chemistry) de Peter Atkins et de Julio de Paula, incorpore les
remaniements et les nouveautés de la 9eédition en langue
anglaise.
Chimie physique de Atkins est un ouvrage fondamental qui,
depuis sa première édition en 1978, a exercé une
influence prépondérante sur le développement et l’en-
seignement de la chimie physique dans le monde. Par une
présentation originale fondée, d’une part, sur l’explication
raisonnée des phénomènes physiques de la matière, et,
d’autre part, sur les liens qui mettent la chimie physique
en amont des autres disciplines plus spécialisées de la
chimie, de la biochimie, des sciences de l’environnement,
etc, l’œuvre de Peter Atkins et de Julio de Paula s’est avérée
essentielle au cours de ces dernières années comme
ouvrage d’accompagnement des cours de chimie et
comme traité général donnant à tous les scientifiques une
vue complète et précise de la chimie physique, science
ardue mais primordiale.
La nouvelle organisation de la 4eédition
Dans cette nouvelle édition, on peut signaler en particulier
une réduction du nombre de chapitres traitant de la
thermodynamique classique et l’existence d’un nouveau
chapitre intitulé « Catalyse ». Un progrès important réside
dans l’incorporation de textes intercalaires, très clairs,
intitulés « Bases mathématiques » qui s’avèreront parti-
culièrement utiles aux étudiants pour bien comprendre le
développement des équations. Les auteurs sont conscients
qu’une bonne connaissance des outils mathématiques per-
met d’éviter bien des difficultés. On retrouve dans
ces « Bases mathématiques » remarquables l’esprit de con-
cision et de synthèse qui caractérise les auteurs.
Le niveau mathématique requis pour les étudiants est
normalement celui des premières années universitaires.
Les atouts de cette édition
Dans la 4eédition, les points clés des différents développe-
ments sont présentés en tête de section. Les précieuses
séries d’exercices et de problèmes, et leurs solutions qui se
trouvent à la fin de l’ouvrage, constituent un apport essen-
tiel pour les étudiants. Les rappels de physique sont présen-
tés dans le chapitre « Fondamentaux » au début de l’ou-
vrage avec un rappel des notions fondamentales de la
chimie.
Comme toujours, les auteurs ont le souci d’adopter un
vocabulaire rigoureux, fondé sur les recommandations de
l’Union Internationale de Chimie Pure et Appliquée
(UICPA).
Traduction de la 9eédition anglaise
Jean Toullec, directeur de recherche honoraire au CNRS,
Université de Versailles Saint-Quentin-en-Yvelines, est
président de la commission spécialisée de terminologie et
de néologie de la chimie et des matériaux (CSTN chimie et
matériaux) qui s’inscrit dans le dispositif d’enrichissement
de la langue française. Il est l'un des coauteurs de
Grandeurs, unités et symboles de la chimie physique, la
traduction française officielle du Green Book de l'Union
internationale de chimie pure et appliquée (UICPA).
Monique Mottet, ingénieur chimiste, est la traductrice de
plusieurs ouvrages de chimie parus aux éditions De Boeck
Supérieur. Elle est l'un des coauteurs de Grandeurs, unités
et symboles de la chimie physique, la traduction française
officielle du Green Book de l'Union internationale de chimie
pure et appliquée (UICPA).
Chimie Physique
Atkins
I
De Paula
Chimie Physique
Atkins
I
De Paula
aDes exercices interactifs à réaliser avec le site
http://global.oup.com/uk/orc/chemistry/pchem9e/
aListe de points clés en tête de section
aDe nombreux exemples d’application
aSéries d’exercices et de problèmes avec leurs solutions
aRappels de physique, chimie et mathématiques
782804 1665199
ISBN : 978-2-8041-6651-9
image : © Steve Morvay - Fotolia.com
ATKINSCHIPHY
Atkins
I
De Paula
Chimie Physique
Conception graphique : P rimo& Primo
www.deboeck.com
4eédition
Traduction de l’anglais par
Jean Toullec et Monique Mottet
ATKINSCHIPHY_chimie inorganique 07/05/13 16:10 Page1

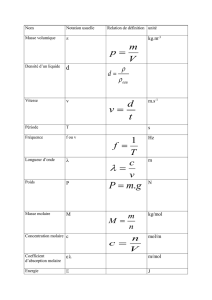
Relations utiles
À 298,15 K
RT 2,4790 kJ ⋅ mol−1
RT/F 25,693 mV
RT ln 10/F 59,160 mV
kT/hc 207,226 cm−1
kT/e 25,693 meV
Vm
° 2,4790 × 10−2 m3 ⋅ mol−1 = 24,790 dm3 ⋅ mol−1
Unités importantes
1 N 1 kg ⋅ m ⋅ s −2 1 J 1 kg ⋅ m 2 ⋅ s −2
1 Pa 1 kg ⋅ m −1 ⋅ s −2 1 W 1 J ⋅ s −1
1 V 1 J ⋅ C −1 1 A 1 C ⋅ s −1
1 T 1 kg ⋅ s −2 ⋅ A−1 1 P 10−1 kg ⋅ m −1 ⋅ s −1
1 S 1 Ω−1 =1 A ⋅ V −1
Facteurs de conservation des unités
q/°C = T/K − 273,15*
1 eV 1,602 18 × 10−19 J
96,485 kJ ⋅ mol−1
8 065,5 cm−1
1 cal 4,184* J
1 atm 101,325* kPa
760* Torr
1 cm−1 1,9864 × 10−23 J
1 D 3,335 64 × 10−30 C ⋅ m
1 Å 10−10 m*
(*Valeurs exactes)
Relations mathématiques
π = 3,141 592 653 59 . . .
e = 2,718 281 828 46 . . .
Logarithmes et exponentielles
ln x + ln y + . . . = ln xy . . .
ln x − ln y = ln (x/y)
a ln x = ln xa
ln x = (ln 10) log x = (2,302 585 . . .) log x
exeyez . . . = ex + y + z + . . .
ex/ey . . . = ex − y
(ex)a = eax
e±ix = cos x ± i sin x
Préxes
z a f p n mm c d da k M G T P
zepto atto femto pico nano micro milli centi déci déca kilo méga giga téra péta
10–21 10–18 10–15 10–12 10–9 10–6 10–3 10–2 10–1 1011031061091012 1015
Développement de Taylor
ƒ(x) =
a
(x−a)n
ex=1 +x +x2+...
ln x =(x −1) −(x −1)2+(x −1)3−(x −1)4+...
ln(1 +x) =x −x2+x3...
=1 −x +x2...
1
1 +x
1
3
1
2
1
4
1
3
1
2
1
2
D
E
F
dnf
dxn
A
B
C
1
n!
∞
∑
n=0
Dérivées
d(f +g) =df +dg
d(fg) =fdg +gdf
d=df−dg
=
zyx
=−1
(∂y/∂x)z=1/(∂x/∂y)z
=nxn−1
eax =aeax
=1
x
dlnx
dx
d
dx
dxn
dx
∂z
∂y
∂x
∂z
∂y
∂x
dg
dt
df
dg
df
dt
f
g2
1
g
f
g
Intégrales
xndx=+constante
dx=ln x+constante
∞
0
xne−axdx=
sin2axdx=x−sin 2ax +constante
sin axsin bxdx=−+constante
si a2≠b2
erf z=
z
0
e−y2dy
erfc z =1 −erf z
2
π1/2
sin(a + b)x
2(a + b)
sin(a − b)x
2(a − b)
1
4a
1
2
n!
an+1
1
x
xn+1
n +1

Chimie physique
ATKINSCHIPHY-pgeTitre.indd 1 6/05/13 09:15

Chez le même éditeur
ATKINS P.W. et JONES L., Principes de chimie, 2e édition
ATKINS P.W., Au cœur des réactions chimiques
ATKINS P.W., Les 4 grands principes qui régissent l‘univers
BLIEFERT C., PERRAUD R., Chimie de l’environnement. Air, eau, sol, déchets, 2e édition
CLAYDEN J., GREEVES N., WARREN S., WOTHERS P., Chimie organique
MCQUARRIE D.A., GALLOGLY E.B., ROCK P.A., Chimie générale, 3e éd.
MURRAY R.K., BENDER D.A., BOTHAM K.H., KENNELLY P.J., RODWELL V.W., WEIL A., Biochimie de Harper, 5e édition
SHRIVER D.F., ATKINS P.W., Chimie inorganique
SILVERSTEIN R.M., WEBSTER F.X., KIEMLE D.J., Identification spectroscopique de composés organiques, 2e édition
VOLLHARDT P.C.K., SCHORE N.E., Traité de chimie organique, 5e édition
ATKINSCHIPHY-pgeTitre.indd 2 6/05/13 09:15

Chimie physique
4e édition
Traduction de la 9e édition anglaise par Jean Toullec et Monique Mottet
Peter Atkins | Julio De Paula
ATKINSCHIPHY-pgeTitre.indd 3 6/05/13 09:15
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
1
/
96
100%