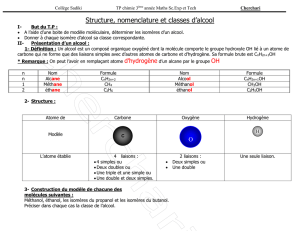
UE CHI363 Chimie industrielle

Chapitre 3 : Cohésion d’un échantillon
de matière et liaisons intermoléculaires
1
A. Distributions de charges dans les atomes et molécules
•Moments dipolaires permanents
•Moments dipolaires instantanés
B. Définition de différents types de liaisons intermoléculaires
•Forces de van der Waals : liaisons non spécifiques et non
saturables
•Pont Hydrogène : liaison spécifique et saturable
C. Mise en évidence : manifestations expérimentales
•Températures de changements d’états (fusion, ébullition)
•Miscibilités de deux corps ou mise en solution d’un corps dans
un solvant
•Réactivité chimique (influence du milieu réactionnel sur la
vitesse d’une réaction, sur la position d’un équilibre)

2
Distributions de charges et dipôles électriques
Un « dipôle électrique » est constitué de deux charges ponctuelles q et
q’ = -q situées à une distance d en deux points M et M’ ; la séparation
des charges est décrite par un vecteur m appelé « moment dipolaire
électrique » m = q.d.u, où d indique la distance, q indique la charge et u
est un vecteur unitaire de la direction M’M
Pour l’orientation du vecteur m , deux conventions existent :
•orientation du pole négatif vers le pole positif (M’ vers M : plutôt la
convention des physiciens)
•orientation du pole positif vers le pole négatif (M vers M’ : plutôt la
convention des chimistes)
Il est donc recommandé de toujours préciser les signes des charges aux
extrémités du dipôle
en M' , -q
q , en M
m
d

3
Liaisons polaires et moments dipolaires de liaison
Entre deux atomes liés d’électronégativités différentes, le nuage
électronique de liaison est réparti de façon non symétrique : il est attiré
plus fortement vers l’atome le plus électronégatif, créant une séparation
de charges
Une telle liaison est dite liaison polaire et peut être assimilée à un dipôle
électrique (une charge partielle positive du côté de l’atome le moins
électronégatif et une charge partielle négative du côté de l’atome le plus
électronégatif) ; elle est ainsi caractérisée par son moment dipolaire, de
norme m = q.d
•q et -q sont les charges partielles, localisées aux noyaux, traduisant
la dissymétrie du nuage électronique de liaison
•d est la distance internucléaire d’équilibre
•la direction du vecteur est l’axe nucléaire
Unité usuelle : le Debye, 1 D = 1/3.10-29 C.m ; le moment dipolaire d’un
dipôle constitué de deux charges -e et +e distantes de 10-10 m a pour
valeur 4,8 D.

4
Electronégativité et polarité des liaisons
•Electronégativité d’une OA : lorsqu’une OA de valence d’un atome
A est plus stable qu’une OA de valence d’un atome B, elle est dite
plus électronégative
•Electronégativité d’un atome considéré globalement : de façon
qualitative, elle caractérise l’aptitude à stabiliser les électrons ; il
existe plusieurs échelles pour la quantifier : cette année, les
valeurs utilisées sont celles de l’échelle de Pauling
•Remarque : l’électronégativité d’un atome dépend de son
environnement dans un édifice
•Identification de liaisons polaires : il s’agit de comparer les
électronégativités des atomes liés => double conclusion :
•le caractère polaire d’une liaison
•le sens de la dissymétrie de la répartition du nuage
électronique
•Exemples de liaisons polaires : H-O ; H-N

5
Le « pourcentage de caractère ionique » (ou pourcentage d’ionicité)
d’une liaison polaire est le rapport de la charge q portée par les pôles du
dipôle à la charge élémentaire e : c’est une description de la liaison
polaire par comparaison à une référence qui est la limite ionique
Pourcentage d’ionicité d’une liaison
m expérimental
%ionicité = F H
-e
d
liaison 100% ionique: F-H+
m ionique
=
m expérimental x 100
e.d
+e
données expérimentales
m
Espèce HF HCl HBr HI
d / pm 92 127 141 161
m / D 1,83 1,08 0,82 0,44
E / kJ.mol-1
565 431 366 299
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
1
/
26
100%