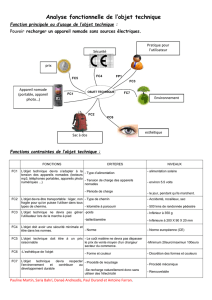
Etude du récepteur pro-apoptotique FAS dans des cellules

UNIVERSITE DE LIMOGES
Faculté des Sciences
Ecole Doctorale ED 258
UMR-CNRS 6101- Laboratoire de Neuro-immunologie
N° 20 - 2003
THESE
Pour obtenir le grade de
DOCTEUR DE L’UNIVERSITE DE LIMOGES
Discipline : SCIENCES DE LA VIE ET DE LA SANTE
Option : IMMUNOLOGIE, NEUROSCIENCE
Présentée et soutenue publiquement par
Christophe LAUTRETTE
Le 1
er
Juillet 2003
ETUDE DU RÉCEPTEUR PRO-APOPTOTIQUE FAS
DANS DES CELLULES NEURONALES ET
LYMPHOCYTAIRES T HUMAINES
Directeur de Thèse : Pr. Marie-Odile JAUBERTEAU-MARCHAN
Dr C. AYER-LE LIEVRE, DR-CNRS (Université de Limoges) PRESIDENT
Dr N. BAUMANN, DR - INSERM (Université de Paris VI) RAPPORTEUR
Dr JC. LECRON, MCU-PH (Université de Poitiers) RAPPORTEUR
Pr MH. RATINAUD, PU (Université de Limoges) EXAMINATEUR
Pr MO. JAUBERTEAU-MARCHAN, PU-PH (Université de Limoges) DIRECTEUR

A mes parents, mes grands-parents, mon frère et ma sœur,
A Caroline,

Remerciements
Nous exprimons toute notre reconnaissance au professeur Marie-Odile Jauberteau-
Marchan qui est à l’origine de cette étude réalisée sous sa direction. Ses compétences
scientifiques et sa constante disponibilité nous ont permis de mener à bien ce travail. Nos
sincères remerciements lui sont adressés.
Nous sommes très sensibles à l’honneur que nous fait le Docteur Nicole Baumann en
acceptant de juger ce travail et nous lui exprimons toute notre reconnaissance et notre profond
respect.
Nous exprimons tous nos remerciements au Docteur Jean-Claude Lecron qui nous fait
l’honneur de juger ce travail et nous lui témoignons toute notre reconnaissance et notre
profond respect.
Nous remercions sincèrement le Docteur Christiane Ayer-Le Lièvre qui nous fait
l’honneur de présider ce jury. Nous lui exprimons notre profond respect.
Nous remercions vivement le professeur Marie-Hélène Ratinaud d'avoir accepté de
participer à ce jury. Nous lui témoignons toute notre reconnaissance.

Je remercie vivement la société Diaclone de m’avoir fourni gracieusement les
anticorps nécessaires à cette étude. Mes remerciements s’adressent plus particulièrement à M.
J. Widjenes et à Mme C. Vermot-Desroches qui ont toujours soutenu ces travaux. J’espère
sincèrement que les publications issues des ces études contribueront à mieux faire connaître
l’excellence de leurs réactifs.
Je remercie aussi tout particulièrement le Conseil Régional du Limousin pour son
soutien financier au cours de cette étude.
Je remercie aussi vivement l’école doctorale Science - Technologie – Santé, toujours à
l’écoute de nos problèmes doctoraux, et dont les formations ont été très enrichissantes. Je
remercie tout particulièrement Mr Vareille, directeur de ED-258, et Melle Gaëlle Peyrat.
Merci à tout notre laboratoire. A Stéphanie pour l’aide apportée dans ce travail, sa
gentillesse, et sa patience devant nos concepts personnels du rangement. Je remercie aussi
vivement Bertrand pour son aide, son soutien, et ses conseils ; une grande partie de ces
travaux a été soutenue par ses qualités. Merci également à Elodie, pour l’aide qu’elle m’a
apportée dans cette étude, mais aussi pour sa grande gentillesse et sa constante bonne humeur.
Je tiens aussi à remercier Murielle pour son aide et ses compétences scientifiques précieuses
qu’elle a toujours partagées; et pour sa gentillesse et sa disponibilité. Je remercie vivement le
Dr Caroline Boda pour sa gentillesse, son écoute, et son aide indispensable à l’étude et aux
soins de mes souris.
Je remercie beaucoup également Caroline et Cathy. Toujours à notre écoute, les avoir
comme voisines fut un grand plaisir.
Je remercie vivement le laboratoire de Mme Ayer-Le Lièvre avec qui nous avons
souvent travaillé : Fafa ; Isabelle pour son aide précieuse dans mes travaux, mais surtout pour
son amitié très importante dans la réalisation de cette étude ; et Barbara, toujours prête à
rendre service.
Je remercie aussi le laboratoire de Michel Cogné qui m’a apporté son aide à diverses
occasions. Je citerai tout particulièrement Krap, Florence, Zelia, et Hei Lanne...
Une petite ligne à part pour Laurence qui m’a bien souvent aidé, tant pour des conseils
techniques que pour retrouver des réactifs perdus… Merci surtout pour ta constante bonne
humeur communicative.

Merci aussi à toute l’équipe de Marie-Hélène Ratinaud, de Danielle Troutaud et de
Mireille Verdier. Un petit mot à Thomas, pour toutes nos excellentes discussions sur
l’apoptose et autres.
Merci également au laboratoire du Pr Cardot et à Serge Battu pour son aide et ses
conseils. Travailler ensemble fut pour moi très enrichissant.
Mes remerciements à Christophe Lejeune pour son aide et la bonne humeur
quotidienne qu’il apporta au laboratoire. Merci aussi à Dominique qui nous permet de
travailler dans des locaux toujours propres.
Merci aussi à Sophie Collot pour sa gentillesse et son aide à diverses occasions.
Je souhaite également remercier le personnel des Laboratoires d'Immunologie et
d’Anatomo-Pathologie du CHU de Limoges pour avoir facilité ce travail. Je remercie tout
particulièrement le Dr Mireille Drouet pour son aide en cytométrie en flux et le Pr F.
Labrousse pour les études histopathologiques des souris.
Ces remerciements s’adressent également au personnel du service de gynécologie
obstétrique, sans l’aide duquel une partie importante de ces travaux n’aurait pas pu être
réalisée.
De nombreux remerciements, pour leur aide indispensable et leur constante
disponibilité, sont adressés à Jacques, de l'animalerie du CHU ; ainsi qu’à Florence et Sylvie,
de l’animalerie de la Faculté de médecine. Merci aussi à Monsieur Herblot pour son aide dans
mes travaux photos.
Merci enfin à tous mes amis : Anne, Yann, Marie, Thomas, Corinne, Fred, Sylvie,
Valérie, Stéphanie et Gilles.
Mes derniers remerciements s’adressent à toute ma famille. Je remercie tout
particulièrement Sophie, ma sœur, Alexis, mon frère, mes parents et mes grands-parents, qui
m’ont toujours aidé, soutenu et encouragé au cours de mes études et, bien évidemment, de
cette thèse qui sans leur soutien n’aurait pu être réalisée. Merci enfin à Caroline pour son aide
et son soutien inconditionnel.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
1
/
211
100%