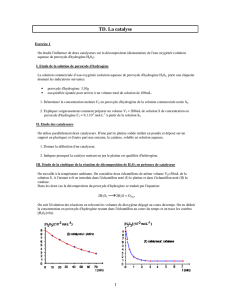
Pd/ZrO2-SO4 - Université De Strasbourg

C
CC
C
C
CC
CA
AA
A
A
AA
AR
RR
R
R
RR
RA
AA
A
A
AA
AC
CC
C
C
CC
CT
TT
T
T
TT
TE
EE
E
E
EE
ER
RR
R
R
RR
RE
EE
E
E
EE
E
B
BB
B
B
BB
BI
II
I
I
II
IF
FF
F
F
FF
FO
OO
O
O
OO
ON
NN
N
N
NN
NC
CC
C
C
CC
CT
TT
T
T
TT
TI
II
I
I
II
IO
OO
O
O
OO
ON
NN
N
N
NN
NN
NN
N
N
NN
NE
EE
E
E
EE
EL
LL
L
L
LL
L
P
PP
P
P
PP
PA
AA
A
A
AA
AR
RR
R
R
RR
RT
TT
T
T
TT
TI
II
I
I
II
IC
CC
C
C
CC
CU
UU
U
U
UU
UL
LL
L
L
LL
LI
II
I
I
II
IE
EE
E
E
EE
ER
RR
R
R
RR
R
D
DD
D
D
DD
DE
EE
E
E
EE
ES
SS
S
S
SS
S
C
CC
C
C
CC
CA
AA
A
A
AA
AT
TT
T
T
TT
TA
AA
A
A
AA
AL
LL
L
L
LL
LY
YY
Y
Y
YY
YS
SS
S
S
SS
SE
EE
E
E
EE
EU
UU
U
U
UU
UR
RR
R
R
RR
RS
SS
S
S
SS
S
P
PP
P
P
PP
Pt
tt
t
t
tt
t,
,,
,
,
,,
,
I
II
I
I
II
Ir
rr
r
r
rr
r,
,,
,
,
,,
,
O
OO
O
O
OO
OU
UU
U
U
UU
U
P
PP
P
P
PP
Pd
dd
d
d
dd
d
S
SS
S
S
SS
SU
UU
U
U
UU
UP
PP
P
P
PP
PP
PP
P
P
PP
PO
OO
O
O
OO
OR
RR
R
R
RR
RT
TT
T
T
TT
TE
EE
E
E
EE
ES
SS
S
S
SS
S
S
SS
S
S
SS
SU
UU
U
U
UU
UR
RR
R
R
RR
R
Z
ZZ
Z
Z
ZZ
ZI
II
I
I
II
IR
RR
R
R
RR
RC
CC
C
C
CC
CO
OO
O
O
OO
ON
NN
N
N
NN
NE
EE
E
E
EE
E
S
SS
S
S
SS
SU
UU
U
U
UU
UL
LL
L
L
LL
LF
FF
F
F
FF
FA
AA
A
A
AA
AT
TT
T
T
TT
TE
EE
E
E
EE
EE
EE
E
E
EE
E
D
DD
D
D
DD
DA
AA
A
A
AA
AN
NN
N
N
NN
NS
SS
S
S
SS
S
L
LL
L
L
LL
LE
EE
E
E
EE
ES
SS
S
S
SS
S
R
RR
R
R
RR
RE
EE
E
E
EE
EA
AA
A
A
AA
AC
CC
C
C
CC
CT
TT
T
T
TT
TI
II
I
I
II
IO
OO
O
O
OO
ON
NN
N
N
NN
NS
SS
S
S
SS
S
D
DD
D
D
DD
DE
EE
E
E
EE
E
R
RR
R
R
RR
RE
EE
E
E
EE
EF
FF
F
F
FF
FO
OO
O
O
OO
OR
RR
R
R
RR
RM
MM
M
M
MM
MA
AA
A
A
AA
AG
GG
G
G
GG
GE
EE
E
E
EE
E
D
DD
D
D
DD
D’
’’
’
’
’’
’A
AA
A
A
AA
AL
LL
L
L
LL
LC
CC
C
C
CC
CA
AA
A
A
AA
AN
NN
N
N
NN
NE
EE
E
E
EE
ES
SS
S
S
SS
S
Présentée et Soutenue publiquement le 25
2525
25-
--
-06
0606
06-
--
-2002
20022002
2002
M
MM
MEMBRES DU JURY
EMBRES DU JURYEMBRES DU JURY
EMBRES DU JURY
Rapporteur Interne : M. Je
M. JeM. Je
M. Jean SOMMER
an SOMMERan SOMMER
an SOMMER
Rapporteur Externe : Mme Christine TRAVERS
Mme Christine TRAVERSMme Christine TRAVERS
Mme Christine TRAVERS
Rapporteur Externe : M. Jean
M. JeanM. Jean
M. Jean-
--
-Pierre GILSON
Pierre GILSONPierre GILSON
Pierre GILSON
Examinateur : M. Gilbert MAIRE
M. Gilbert MAIREM. Gilbert MAIRE
M. Gilbert MAIRE
Membre invité : M.
M. M.
M. Georges HADZIIOANNOU
Georges HADZIIOANNOUGeorges HADZIIOANNOU
Georges HADZIIOANNOU
Directeur de Thèse : M. François GARIN
M. François GARINM. François GARIN
M. François GARIN
LABORATOIRE DES MATERIAUX SURFACES ET PROCEDES POUR LA CATALYSE (LMSPC)
UMR7515 CNRS, ECPM, UNIVERSITE LOUIS PASTEUR – 25 rue Becquerel, F-67083 Strasbourg Cedex
THESE
THESETHESE
THESE
présentée pour obtenir le grade de
DOCTEUR
DOCTEURDOCTEUR
DOCTEUR
Discipline : C
CC
CHIMIE PHYS
HIMIE PHYSHIMIE PHYS
HIMIE PHYSIQUE
IQUEIQUE
IQUE
par U
UU
U
U
UU
Um
mm
m
m
mm
mi
ii
i
i
ii
it
tt
t
t
tt
t
B
BB
B
B
BB
Bi
ii
i
i
ii
il
ll
l
l
ll
lg
gg
g
g
gg
ge
ee
e
e
ee
e
D
DD
D
D
DD
DE
EE
E
E
EE
EM
MM
M
M
MM
MI
II
I
I
II
IR
RR
R
R
RR
RC
CC
C
C
CC
CI
II
I
I
II
I

Thèse de doctorat en Chimie physique Umit Bilge DEMIRCI
1999·2002
- 2 -

Thèse de doctorat en Chimie physique Umit Bilge DEMIRCI
1999·2002
- 3 -
Babama - A mon Père
Anneme
-
A ma Mère
Kardeslerime - A mon Frère et a ma soeur
Esime -
--
- A mon épouse

Thèse de doctorat en Chimie physique Umit Bilge DEMIRCI
1999·2002
- 4 -

Thèse de doctorat en Chimie physique Umit Bilge DEMIRCI
1999·2002
- 5 -
A
AA
A
A
AA
AV
VV
V
V
VV
VA
AA
A
A
AA
AN
NN
N
N
NN
NT
TT
T
T
TT
T-
--
-
-
--
-P
PP
P
P
PP
PR
RR
R
R
RR
RO
OO
O
O
OO
OP
PP
P
P
PP
PR
RR
R
R
RR
RO
OO
O
O
OO
OS
SS
S
S
SS
S
-
--
-
-
--
-
R
RR
R
R
RR
RE
EE
E
E
EE
EM
MM
M
M
MM
ME
EE
E
E
EE
ER
RR
R
R
RR
RC
CC
C
C
CC
CI
II
I
I
II
IE
EE
E
E
EE
EM
MM
M
M
MM
ME
EE
E
E
EE
EN
NN
N
N
NN
NT
TT
T
T
TT
TS
SS
S
S
SS
S
Mes trois années de Doctorat se sont déroulées au Laboratoire des Matériaux,
Surfaces, et Procédés pour la Catalyse (LMSPC), anciennement Laboratoire d’Etudes de la
Réactivité Catalytique des Surfaces et Interfaces (LERCSI) sous la direction du Docteur
François GARIN.
Je profite de ces pages « libres et personnelles » pour lui exprimer ma profonde
gratitude, d’une part pour m’avoir accueilli dans son laboratoire, et d’autre part, pour m’avoir
permis de travailler sur un sujet des plus intéressant. Je tiens aussi à le remercier pour sa
disponibilité, ses suggestions, ses conseils et pour nos nombreuses discussions qui nous ont
permis de mener à bien nos recherches. Merci enfin pour la confiance et la liberté d’initiative.
Mes remerciements vont également à Madame Christine TRAVERS (ENSPM-IFP,
Rueil-Malmaison), Monsieur Jean SOMMER (Université Louis Pasteur, Strasbourg),
Monsieur Jean-Pierre GILSON (Université de Caen, Caen), Monsieur Gilbert MAIRE
(Université Louis Pasteur, Strasbourg) et Monsieur Georges HADZIIOANNOU (Université
Louis Pasteur, ECPM, Strasbourg) pour l’honneur qu’ils me font de juger ce travail.
Je tiens à remercier tous ceux qui ont collaboré à l’accomplissement de ce travail, en
particulier à Mme Paule GIRARD, pour les synthèses et analyses des alcanes marqués, Mme
Suzanne LIBS, pour l’installation du chromatographe, M. Pierre BERNHARDT, pour les
installations informatiques et électroniques, M. Jean-Louis SCHMITT, pour les modifications
techniques du bâti pression atmosphérique, M. Michel WOLF, pour le montage du bâti
moyenne pression, M. Georges PONCELET, pour ses catalyseurs qu’il nous a confiés, et, M.
Alain Goeppert, pour m’avoir encadré lors des réactions de dosage des sites acides.
Un grand merci à tous mes collègues de laboratoire (présents et passés) qui ont
contribué à créer une atmosphère conviviale, détendue et joviale, et plus particulièrement, à
Frédéric HOERNEL et à François DI GREGORIO.
Encore merci pour tout et à tous !
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
242
243
244
245
246
247
248
249
250
251
252
253
254
255
256
257
258
259
260
261
262
1
/
262
100%