Conversions de fonctions - Anne CURK

CONVERSION DES FONCTIONS ORGANIQUES : COMPLEMENTS................4
I – CONVERSION DES ALCENES ET ALCYNES.......................................................................4
1-De l'alcène à l'alcool.........................................................................................................4
1-
A
H
YDRATATION ACIDE DES ALCENES
:
ADDITION ELECTROPHILE SOUS CONTROLE CINETIQUE
.....................4
H
+
se fixe --> le C
+
le plus stable => OH est sur le C issu du C
+
le plus stable ( Règle de Markovnikoff )
=> OH n'est pas en bout de chaîne ( car C
+
primaire ) sauf si C
+
en résonance
Choix de l'acide : acide à anion NON nucléophile => H
2
SO
4
privilégié (dilué dans H
2
O )
1-
B
H
YDROBORATION DES ALCENES
:
ADDITION ELECTROPHILE SOUS CONTROLE STERIQUE
...........................6
Contrôle stérique : B de BH
3
se place prioritairement sous le C le moins encombré de la = la moins encombrée =>
OH sur le C le moins encombré de la = la moins encombrée.
Gain de régiosélectivité avec des boranes encombrés : R
2
BH au lieu de BH
3
2-Hydratation des alcynes : de l'alcyne à la cétone..........................................................8
Nécessité d'un catalyseur metallique : Hg
2+
en plus de H
+
--> énol --> cétone après tautomérie ( Hors programme)
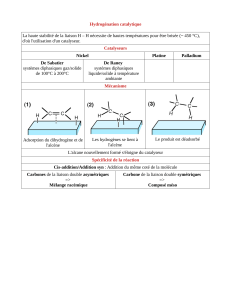
3- Hydrogénation des alcènes et des alcynes (Bilan = Réduction, +H
2
)........................8
3-
A
H
YDROGENATION DES ALCENES ET ALCYNES EN CATALYSE HETEROGENE
................................................9
• Resultats expérimentaux.......................................................................................................................................9
H
2
se fixe sur le catalyseur solide ; SYN addition à P = 1 bar et T ambiante
• Mécanisme ............................................................................................................................................................9
1- Adsorption ( physisorption, puis chimiesorption) ; 2- réaction ; 3- désorption
• Action sur les alcynes..........................................................................................................................................10
Sans poison : alcyne + 2 H
2
--> alcane Avec poison ( encombre le catalyseur ) : alcyne + H
2
--> alcène Z
• Action sur d'autres insaturations .........................................................................................................................10
à P et T ambiantes : NON
En augmentant P et T => réduction des C=O --> CH-OH ou à P et T énormes : benzène --> cyclohexane
• Méthode d'obtention des catalyseurs à base de nickel.......................................................................................10
2 méthodes : oxydation d'un alliage NiAl par OH
-
: seul Al est oxydé en AlO
22-
=> Ni pulvérulent se dépose
réduction de l'oxyde NiO par H
2
=> Ni pulvérulent se forme.
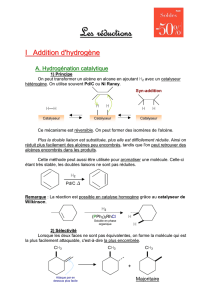
3-
B
H
YDROGENATION DES INSATURATIONS CARBONE CARBONE EN CATALYSE HOMOGENE
...........................12
H
2
+ Catalyseur de Wilkinson = (Rh(PPh
3
)
3
Cl => addition SYN de H
2
sur les alcènes
Catalyseur
+
=> plus rapide (RhL
4+
, PF
6-
) ou (IrL
4+
, PF
6-
)
( on retient catalyseur plan carré à base de Rhodium ou d'iridium )
Régiosélectivité : alcènes peu encombrés et alcènes porteurs d'un groupe attracteur réagissent plus vite.
3-
C
H
YDROGENATIONS ENANTIOSELECTIVES EN CATALYSE HOMOGENE
***...................................................14
Si le Rhodium porte un ligand chiral => induction asymétrique => un seul énantiomère syn obtenu.

1
II- CONVERSION DES FONCTIONS TRIVALENTES...............................................................17
1-Réactivités électrophiles relatives : critère cinétique..................................................17
Niveau BV => Chlorures > anhydrides > thioester > esters > amides > acides carboxyliques
=> réactions spontanées des chlorures, anhydrides et thioesters
=> activation nécessaire pour esters, amides et acides
2- Critère Thermodynamique............................................................................................18
Stabilité gpe partant ie pKa du couple (Hgpe partant / gpe partant) le plus faible ( base = gpe partant la plus stable)
=>Chlorures > anhydrides > thioesters > acides carboxyliques > esters > amides
=>Chlorures obtenus par un réactif spécifique sans utiliser la réactivité électrophile
=>Anhydrides obtenus à partir des chlorures ou voie spécifique hors électrophilie
=>Esters et amides obtenus à partir de tous les autres ( avec ou sans activation selon le critère cinétique)
3-Les
CHLORURES ET ANHYDRIDES
précurseurs privilégiés des esters et amides ............18
3-
A
P
RESENTATION ET SYNTHESE DES CHLORURES ET ANHYDRIDES
............................................................19
Chlorures : RCOOH + SOCl
2
--> RCOCl + SO
2
+ HCl ( idem ROH + SOCl
2
--> RCl + SO
2
+ HCl )
Anhydrides : RCOCl + R'COOH --> R(CO)O(CO)R' + HCl ou 2 RCOOH -- P
4
O
10
ou P
2
O
5
--> R(CO)O(CO)R
dissymétrique très efficace --> anhydride cyclique
3-
B
D
ES CHLORURES D
’
ACYLE AUX ESTERS ET AUX THIOESTERS
.................................................................19
Mécanisme = addition / élimination - pas de catalyse
RCOCl + R'OH --> RCOOR' + HCl ( base non nucléophile pour éliminer HCl )
RCOCl + R'SH --> RCOSR' + HCl ( base non nucléophile pour éliminer HCl )
3-
C
D
ES ANHYDRIDES D
’
ACIDES AUX ESTERS OU AUX THIOESTERS
..............................................................20
Mécanisme = addition / élimination - pas de catalyse
R(CO)O(CO)R + R'OH --> RCOOR' + RCOOH ( pas de base : RCOO- libéré fait office )
R(CO)O(CO)R + R'SH --> RCOSR' + RCOOH ( pas de base : RCOO- libéré fait office )
3-
D
D
ES CHLORURES ET ANHYDRIDES D
’
ACYLE AUX AMIDES
........................................................................20
Mécanisme = addition / élimination - pas de catalyse
RCOCl + ammoniac ou amine primaire ou amine secondaire --> amide + Cl
-
+ H
+
fixé sur base
( pas d'amines tertiaires = base ou assistant nucléophile seulement)
R(CO)O(CO)R' + ammoniac ou amine primaire ou amine secondaire --> amide + Cl
-
+ H
+
fixé sur base
(Voies royales pour l'obtention des amides).

2
4- Synthèses des esters et des amides à partir des acides carboxyliques..................23
4-
A
S
YNTHESE DES ESTERS A PARTIR D
'
UN ACIDE CARBOXYLIQUE
:
NECESSAIRE ACTIVATION
.......................23
• Résultats expérimentaux sans catalyse :............................................................................................................23
Bilan : RCOOH + R'OH = RCOOR' + H
2
O athermique, équilibrée, trop lente
accélérée par chauffage ( reflux ), catalyseur H
+
déplacée par Dean-Stark pour éliminer l'eau formée.
• Activation in situ par H
+
: Mécanisme..................................................................................................................23
1° étape : H
+
sur O du C=O puis addition / élimination ( via prototropie )
• Activation in situ par formation d'anhydride mixte : Mécanisme de l'estérification de Yamaguchi (1979)..........24
Deux étapes : 1) RCCOH + chlorure d'acyle + amine tertiaire (base) 2) + R'OH + DMAP --> RCOOR'
1° activation électrophile par transformation acide --> anhydride
2° régiosélectivité et assistance nucléophile par DMAP
• Activation en biologie ( in vivo ) : rôle de la coenzyme A ( HSCoA ) ..................................................................36
Lecture : activation in situ : acide --> esters phosphorés ou thioesters --> amides
4-
B
- LES AMIDES
A PARTIR DES ACIDES CARBOXYLIQUES
:
REACTION
A/B
PREPONDERANTE
.....................26
SOUCI : l'activité basique des amines est PLUS RAPIDE que l'activité nucléophile des amines.
• Activation en biologie ( in vivo ) : rôle de la coenzyme A ( HSCoA ) ..................................................................26
COOH --> COO
-
par un groupe base de l'enzyme => pb A/B éliminé.
COO
-
nucléophile --> thioester
Thioester + amine --> amide
• Exploitation de la réaction A/B entre acide et amine pour la séparation d’énantiomères...................................27
RCOOH (racémique) + NH
2
R' ( 1 seul énantiomère ) --> RCOO
-
, NH
3+
-R' ( sel = 2 disastéréoisomères )
( non séparables ) (séparables)
• Deshydratation par chauffage du carboxylate d'ammonium : obtention d'amide et de polyamide.....................27
RCOOH + NH
2
R' --> RCOO
-
, NH
3+
-R' --chauffage--> RCOONH-R' + H
2
O ( ne pas proposer au laboratoire)
Courant en industrie et polymérisation ( --> polyamides )
4-
C
- C
AS PARTICULIER DE LA SYNTHESE PEPTIDIQUE
..................................................................................28
Problématique : faire réagir 2 acides aminés entre eux : 2 amides possibles => comment orienter vers l'un ?
Succession de protection / activation / réaction / déprotection pour coupler la bonne fonction amine avec la bonne
fonction acide - Utilisation de résines.
III- HYDROLYSE DES DERIVES D’ACIDE ...............................................................................29
Bilans : RCO - A + H
2
O --> RCOOH + HA ou RCO - A + OH
-
--> RCOO
-
+ HA
Chlorures et anhydrides : rapide, aucune catalyse nécessaire
Mécanisme : addition / élimination
Esters : ●catalyse H
+
: hydrolyse en équilibre déplacée par excès d'eau + lente que celle des amides
Mécanisme : 1°) action de H
+
sur O de C=O puis addition / élimination via prototropie
●+ OH - : hydrolyse basique ou saponification : totale et plus rapide que celle des amides
Mécanisme : addition / élimination
Amides : ●catalyse H
+
: totale et + rapide que celle des esters
Mécanisme : 1°) action de H
+
sur O de C=O puis addition / élimination via prototropie
+ OH
-
: hydrolyse basique totale et + lente que cette des esters .
Mécanisme : addition / élimination
Nitriles : hydrolyse --> amide --> acide carboxylique.

3
IV- CONVERSIONS PAR OXYDOREDUCTION .......................................................................32
1-Réduction des esters en alcools primaires..................................................................32
Mécanisme d'action de Li
+
, AlH
4-
: Al tétravalent, <=> H
-
1°) addition nucléophile de H
-
2°) élimination de RO
-
3°) addition de H
-
sur cétone
puis hydrolyse --> alcool primaire
(analogie avec la double addition d'un organomagnésien sur un ester )
2- Le DIBAL-H : réduction ménagée des esters en aldéhyde.........................................32
2-
A
- S
TRUCTURE ET REACTIVITE
...............................................................................................................32
Al trivalent <=> lacune électronique et H
-
2-
B
- R
EDUCTION PAR LE
DIBAL-H............................................................................................................33
Mécanisme d'action de (iBu)
2
AlH :
1°) action de la lacune électronique sur O 2)° addition nucléophile de H
-
sur C --> intermédiaire stable à -60°C
=> pas d'élimination avant hydrolyse
=> hydrolyse --> hemiacétal --> obtention d'un aldéhyde.
3- Réduction des acides carboxyliques...........................................................................34
Préliminaire : réaction A/B de H
-
sur -COOH => perte d'une partie des hydrures
=> possible avec LiAlH
4
mais agressif et couteux
=> possible avec NaBH
4
mais couteux
=> on préfère passer via l'ester : RCOOH--> RCOCl --> RCOOR' --Hydrure--> alcool ou aldéhyde
4- Oxydation des alcènes en époxydes : rappels et compléments ...............................34
4-
A
- O
BTENTION DES EPOXYDES
(
RAPPEL
)...............................................................................................34
Action électrophile de MCPBA sur alcène nucléophile ( => alcène les plus riches en électrons + réactifs ) --> 2
époxydes énantiomères.
4-
B
E
TUDE DE L
’
OUVERTURE DES EPOXYDES EN MILIEU BASIQUE OU NUCLEOPHILE
......................................35
H
2
O / OH
-
attaque en anti => diol avec les 2 OH en anti ( stéréospécifique )
tout nucléophile fait idem ( voir organomagnésiens ) + réaction sur le site le moins encombré.
Rappel : diol avec les 2 OH en syn : alcène + OsO
4
+ H
2
O + NMO
H
2
O / H
+
: peut passer par C
+
=> perte de stéréospécificité.

4
CONVERSION DES FONCTIONS ORGANIQUES : COMPLEMENTS
On appelle conversion d'un groupe, toute réaction qui en change la fonction, SANS CHANGER LE NBRE
d'ATOMES DE CARBONE. Ce cours complète les conversions déjà vues en 1° année .
I – CONVERSION DES ALCENES ET ALCYNES
La réactivité des alcènes et alcynes provient de leur nuage π, caractéristique de la double ou triple liaison. Ce
nuage constitue une liaison plus faible que la liaison σ .
1-De l'alcène à l'alcool
Bilan :
Ce bilan est donc une addition d'eau appelée HYDRATATION des alcènes, réaction inverse de la déshydratation
des alcools.
On rappelle que la déshydratation d'un alcool se produit soit par action de H
2
SO
4
concentré à chaud sur les
alcools secondaires et tertiaires, soit par action d'un chlorure de mésyle ( activation ) suivie de l'action de la
soude, sur un alcool primaire, soit par E
1
cb sur un β aldol en milieu basique.
On constate d'après ce bilan très simplifié que si l'alcène n'est pas symétrique, se pose obligatoirement la question
de la régiosélectivité de cette réaction. Il existe deux méthodes d'hydratation des alcènes, qui conduisent à deux
régiosélectivités opposées, ce qui permettra donc de situer la fonction alcool dans la position souhaitée.
1-
A
H
YDRATATION ACIDE DES ALCENES
:
ADDITION ELECTROPHILE SOUS CONTROLE CINETIQUE
L'hydratation d'un alcène se produit en présence du catalyseur H
2
SO
4
dilué dans l'eau , à froid.
+
+
OH2
OH2
OH
HOH
H
OH
HOH
H
+
+
MAJ min
min MAJ
H+
HSO4-
H+
HSO4-
Par ailleurs, on obtient toujours la formation d'alcools racémiques : La réaction ne présente aucune
stéréospécificité.
L'analyse de ces résultats implique la formation d'un carbocation intermédiaire, le plus stable possible : la réaction
est donc sous contrôle CINETIQUE :
R
1
R
2
R
3
R
4
+
OH2
OH
R
1
R
2
R
3
R
4
H
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
1
/
39
100%