Cours de chimie Organique - G

Cours de chimie Organique - G. Dupuis - Lycée Faidherbe de Lille
Acides carboxyliques
Nomenclature
Définitions
Les acides carboxyliques forment une classe de composés caractérisés par la présence du
groupe fonctionnel carboxyle CO2H. Le nom de ce groupe caractéristique rappelle qu'il est
constitué formellement d'un groupe carbonyle CO et d'un groupe hydroxyle OH.
Cependant l'interaction entre ces deux groupes est telle qu'on ne peut pas les considérer
individuellement.
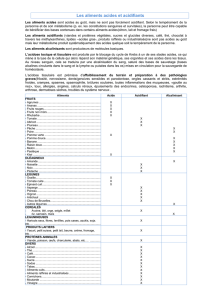
Plusieurs composés sont considérés comme des dérivés d'acides. Les groupes caractéristiques
de ces fonctions dérivées sont donnés dans le tableau ci-dessous.
Acide
Ion carboxylate
Halogénure d'acyle
Anhydride d'acide
Cétène
Ester
Amide
Nitrile
Même si le lien de parenté avec les acides carboxyliques est patent, leurs propriétés chimiques
sont différentes de celles des acides, ce qui justifie une étude séparée.
Acides
On distingue les séries acyclique et cyclique :
Série acyclique
Le nom est formé en ajoutant le suffixe oïque au nom de l'hydrocarbure correspondant
à la chaîne principale. L'atome de carbone du groupe carboxyle porte le numéro 1 qui
n'est pas mentionné.

Acide butanoïque
Acide 3-méthylpentanoïque
Acide hexanedioïque
Acide 3-méthylpent-2-énoïque
Série cyclique
On fait suivre le mot acide du nom de l'hydrocarbure auquel on ajoute le suffixe
carboxylique. L'atome de carbone du carboxyle ne fait pas partie de la chaîne
principale.
Acide benzènecarboxylique
Acide cyclopentanecarboxylique
Ions carboxylate
Les noms des ions carboxylate sont formés à partir de ceux des acides parents en remplaçant
le suffixe oïque par oate ou le suffixe ique par ate.
Pentanoate d'ammonium
Cyclohexanecarboxylate de sodium
Etat naturel
Acides gras
Les acides dont la molécule est constituée d'une longue chaîne d'atomes de carbone sont
appelés acide gras. On peut en effet les obtenir par saponification des graisses qui sont des
esters de ces acides et du glycérol appelés triglycérides.

L'acide hexadécanoïque ou palmitique est un acide gras
possédant 16 atomes de carbone. Le triester qu'il forme avec
le glycérol est la palmitine.
L'acide stéarique constitue un autre exemple d'acide gras qui entre dans la fabrication des
bougies. On trouvera un résumé de ses principales propriétés à la référence [29].
L'acide arachidonique est un acide gras insaturé possédant 20
atomes de carbone.
Il est représenté ci-dessous dans une conformation repliée de la
chaîne. Cela met en évidence la relation entre sa structure et celle
de la prostaglandine PGF2 dont il est un précurseur.
Les prostaglandines sont des acides gras insaturés dont le squelette possède 20 atomes de
carbone. Elles ont un rôle d'hormones locales. La synthèse de la prostaglandine PGF2
représentée ci-dessous a été réalisée par plusieurs équipes. Les grandes étapes de la synthèse
de G. Stork ont été vues dans le chapitre consacré aux composés conjugués.
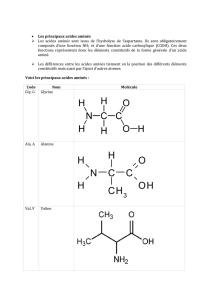
Hydroxy-acides
Les fonctions acide et alcool sont présentes dans de nombreux composés d'origine naturelle.
Ces composés sont des hydroxy-acides.

L'acide mévalonique est un hydroxy-acide qui possède
une grande importance biochimique. C'est en effet le
précurseur d'une grande famille de composés naturels :
les terpènes. Ces composés présents dans les huiles
essentielles des plantes ont une structure dans laquelle
on retrouve la répétition d'unités isoprène.
L'acide cholique a une structure voisine du cholestérol dont il dérive. Les groupes polaires
OH sont situés du même côté de la molécule ce qui permet de distinguer deux faces aux
propriétés physico-chimiques différentes : la face regroupant les groupes polaires est
hydrophile, l'autre est lipophile. En milieu biologique, il est combiné à un acide aminé comme
le glycocolle dans la bile.
L'acide lactique est un hydroxyacide qui possède un intérêt industriel croissant. Il permet la
synthèse d'un lactide dont la polymérisation conduit à un polymère biodégradable, appelé
polylactide (PLA).
Céto-acides
On trouve dans leur molécule un groupe carbonyle et un groupe carboxyle. Le plus important
de ces composés sur le plan biochimique est l'acide pyruvique.
L'acide pyruvique est l'acide 2-oxopropanoïque. Au laboratoire,
on peut le préparer par déshydratation et décarboxylation de
l'acide tartrique.
Acides importants sur le plan industriel

Acide formique
L'acide méthanoïque (formique) est préparé par réaction entre la
soude et le monoxyde de carbone à chaud.
Le traitement par l'acide sulfurique du formiate de sodium fournit
l'acide formique.
Acide acétique
La meilleure préparation industrielle de l'acide éthanoïque est la
carbonylation du méthanol. Ce procédé, mis au point par
Monsanto en 1971, utilise du trichlorure de rhodium comme
catalyseur [23].
Il permet la préparation de plus d'un million de tonnes d'acide
éthanoïque par an. Il sert à préparer notamment l'acétate de vinyle.
Acide téréphtalique
L'acide téréphtalique est produit par oxydation du 1,4-
méthylbenzène par l'oxygène de l'air en présence d'un
catalyseur au cobalt.
Il utilisé dans la synthèse du polyéthylènetéréphtalate.
Propriétés physiques
Acides carboxyliques
Le nom d'un acide carboxylique s'obtient en remplaçant la terminaison e de l'alcane qui
possède le même nombre d'atomes de carbone et en la remplaçant par oïque. Les premiers
termes de la série sont surtout nommés par leur nom trivial qui rappelle leur origine naturelle.
Ces composés ont été parmi les premiers à être identifiés par les chimistes organiciens. Ils
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
1
/
26
100%