Insuffisance surrénalienne

ARTICLE DE REVUE MIG 993
Une maladie dangereuse aux symptômes non spécifiques
Insuffisance surrénalienne
Dr méd. Stefan Fischli
Abteilung für Endokrinologie, Diabetologie und Klinische Ernährung, Departement Medizin, Luzerner Kantonsspital
Dans la pratique clinique quotidienne, il est fréquent de passer à côté du diagnostic
d’insusance surrénalienne en raison des symptômes souvent non spéciques. En
l’absence de traitement, cette maladie est potentiellement fatale. En fonction de sa
cause et de son évolution (aiguë ou chronique), elle se manifeste par diérents
symptômes et anomalies. Cet article de revue a pour objectif de fournir un aperçu
actuel et pertinent pour la pratique de la physiopathologie, des causes, du diagnostic
et du traitement de l’insusance surrénalienne.
Introduction
Plus de 160ans après la première description de l’insuf-
sance surrénalienne primaire par Thomas Addison et
66 ans après l’attribution du prix Nobel à Kendall,
Reichstein et Hench pour leur découverte et première
utilisation clinique de la cortisone, la substitution sur-
rénalienne est encore pratiquée, avec des préparations
pratiquement inchangées depuis des décennies. Des
données récentes montrent l’inuence de la maladie et
de son traitement sur la morbidité et probablement
aussi la mortalité et révèlent qu’avec les modalités thé-
rapeutiques actuelles, nous sommes encore bien loin
d’un traitement de substitution le plus physiologique
possible face à un système complexe et hautement dy-
namique.
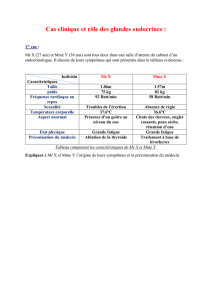
Physiologie et physiopathologie de l’axe
hypothalamo-hypophyso-surrénalien
(g. 1)
Physiologie
Le système hypothalamo-hypophyso-surrénalien (axe
corticotrope) joue un rôle essentiel dans l’adaptation
de l’organisme aux situations de stress. L’activation de
l’axe du cortisol entraîne la mise à disposition de subs-
trats énergiques, une rétention hydrique, l’élévation de
la pression artérielle et du débit cardiaque, ainsi qu’un
contrôle de la réponse immunitaire. Seule l’intégrité
de l’ensemble de l’axe, de pair avec le système nerveux
sympathique, garantit une «réponse au stress» adéquate
et assure ainsi le maintien des fonctions corporelles
essentielles à l’homéostasie [1].
L’hormone de contrôle centrale de l’axe corticotrope
est l’
adrénocorticotrophine
(ACTH)
hypophysaire, qui
est sécrétée de manière pulsatile et circadienne. La sé-
crétion d’ACTH et donc aussi de cortisol atteint son
maximum tôt le matin et son minimum aux alentours
de minuit. Le caractère pulsatile de la sécrétion d’ACTH
semble jouer un rôle central dans toutes les actions
médiées par l’ACTH[2]. L’ACTH est soumise au contrôle
de plusieurs facteurs sécrétagogues, tels que la cortico-
libérine («corticotropin-releasing hormone», CRH) ou
l’hormone antidiurétique (ADH). La liaison de l’ACTH
aux récepteurs de la mélanocortine de type 2 des cellules
adrénocorticales entraîne la stimulation de la synthèse
et de la sécrétion de cortisol et exerce des eets tro-
phiques sur le tissu surrénalien.
L’axe corticotrope peut également être stimulé par des
facteurs indépendants de l’ACTH. En font partie les
Stefan Fischli
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(46):993–1003

ARTICLE DE REVUE MIG 994
cytokines, les adipokines, les substances endothéliales,
les toxines bactériennes et virales, ainsi que l’inner-
vation neuronale directe [1,3].
Le cortisol circulant dans le plasma est lié aux proté-
ines à env. 95% (à la transcortine [«corticosteroid-
binding globulin», CBG] et à l’albumine); seuls env. 5%
se trouvent sous forme libre et peuvent déployer des
eets par liaison au récepteur hormonal. Diérents
états peuvent augmenter ou abaisser la concentration
de protéines de liaison, ce qui entraîne également des
modications des concentrations hormonales totales
déterminées en laboratoire: des concentrations accrues
de CBG sont retrouvées en cas de concentrations élevées
d’œstrogènes (par ex. grossesse, prise de contraceptifs
oraux) ou en cas d’hyperthyroïdie. A l’inverse, l’hypo-
thyroïdie et les décits en protéines (par ex. syndrome
néphrotique, insusance hépatique sévère, malnutri-
tion) abaissent la concentration de CBG.
L’
aldostérone
produite dans la zone glomérulée est régu-
lée de façon primaire par le système rénine-angioten-
sine-aldostérone et de façon secondaire par d’autres
facteurs tels que le potassium sérique. Les androgènes
surrénaliens
déhydroépiandrostérone
(DHEA) et
sulfate
de déhydroépiandrostérone (S-DHEA)
proviennent
de la
zone interne (zone fasciculée/réticulée) de la glande
surrénale, sont soumis au contrôle de l’ACTH et sont
inuencés par l’âge et le sexe.
Physiopathologie
Comme d’autres maladies endocriniennes, l’insusance
surrénalienne est classiée, en fonction du site touché,
en primaire (glande surrénale), secondaire (hypophyse)
Rythmique
Facteurs de stress
Hypothalamus
Angiotensine II
Potassium sérique
Zone interne
Zone fasciculée/réticulée
DHEA/S-DHEA
Cortisol
Zone glomérulée
Aldostérone
Cortisol libre
Pathogènes
Toxines
Glande
surrénale
11 β-HSD2
5α-réductase
5β-réductase
Dégradation
CBG
CBG
CBG
Effets
GR
Tissu
périphérique
Cortisol
lié aux
protéines
Adipocytes
Adipokines
TNF-α
IL-6
ACTH
MC2-R
TR
Glucocorticoïdes
exogènes
Cellules
immunitaires
Hypophyse
CRH ADH
Figure 1: Physiologie/physiopathologie de l’axe hypothalamo-hypophyso-surrénalien et du métabolisme périphérique du cortisol.
Les flèches vertes indiquent les mécanismes pouvant stimuler l’axe corticotrope indépendamment de l’ACTH.
CRH: corticolibérine; ADH: hormone antidiurétique; ACTH: adrénocorticotrophine; MC2-R: récepteur de la mélanocortine de type 2; TR: récepteur de type
Toll; GR: récepteur des glucocorticoïdes; CBG: transcortine; 11βHSD2: 11β-hydroxystéroïde déshydrogénase 2; TNF-α: facteur de nécrose tumorale α;
IL-6: interleukine 6.
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(46):993–1003

ARTICLE DE REVUE MIG 995
et tertiaire (hypothalamus). Les deux dernières formes
sont également désignées par le terme
insusance sur-
rénalienne centrale
. Dans la
forme primaire,
l’ensemble
de la glande surrénale est touchée, aectant ainsi la
sécrétion de glucocorticoïdes, de minéralocorticoïdes
et de DHEA/S-DHEA. En cas de surrénalite auto-immune,
la décience des diérentes hormones est soumise à
une séquence temporelle. La zone glomérulée produi-
sant l’aldostérone est d’abord touchée et la maladie se
caractérise initialement par un faible taux d’aldosté-
rone, des valeurs élevées de rénine, mais une sécrétion
intacte de cortisol [4]. La sécrétion de DHEA est elle
aussi limitée dès le stade préclinique [5]. En cas d’
insuf-
sance surrénalienne centrale,
un décit en CRH ou
ACTH entraîne rapidement une atrophie de la glande
surrénale. La sécrétion de cortisol et des androgènes
surrénaliens s’en trouve perturbée, mais la sécrétion
d’aldostérone reste
toujours
intacte. Dès lors, de faibles
taux de DHEA/S-DHEA constituent également une ano-
malie typique chez les patients sourant d’insusance
surrénalienne centrale [6,7].
Les
aections très graves,
telles que le sepsis, repré-
sentent une situation extrême pour l’axe corticotrope
et elles n’aectent pas uniquement la régulation, mais
également le métabolisme du cortisol et l’action hor-
monale dans les tissus périphériques et au niveau du
récepteur. Des données récentes montrent que chez les
malades critiques, la production de cortisol est unique-
ment augmentée de façon minime, voire pas du tout
[8, 9]. Au cours des premiers jours, la concentration
d’ACTH est accrue, puis elle diminue (phénomène ap-
pelé
dissociation ACTH-cortisol
) [10]. L’un des princi-
paux mécanismes qui assure des concentrations éle-
vées de cortisol sérique dans de telles situations est la
réduction rapide et drastique de la dégradation du cor-
tisol par inhibition des enzymes prévues à cet eet
(11β-hydroxystéroïde déshydrogénase de type 2, 5α- et
5β-réductases) [8]. En outre, d’autres mécanismes adap-
tatifs assurent une disponibilité accrue du cortisol en
périphérie (diminution de la concentration de CBG
avec élévation du cortisol libre, augmentation de l’a-
nité du récepteur des glucocorticoïdes pour le cortisol
par les cytokines) [11]. L’existence d’une
insusance sur-
rénalienne relative
chez les malades critiques («critical
illness-related corticosteroid insuciency», CIRCI),
une dénition homogène de ce concept et les méca-
nismes sous-jacents restent toujours sujets à contro-
verse dans la littérature [12]. Les hypothèses émises
incluent l’inuence des cytokines pro-inammatoires
sur les récepteurs de l’ACTH et des glucocorticoïdes, les
ischémies critiques dans le système hypothalamo-
hypophysaire et la glande surrénale, la suppression des
eets trophiques en raison de faibles taux d’ACTH, les
décits de substrats en raison d’un faible taux de cho-
lestérol et le stress oxydatif [13].
La
crise addisonienne,
en tant que forme extrême du
décit absolu en glucocorticoïdes, illustre remarqua-
blement l’absence d’actions majeures des glucocorti-
coïdes en cas de réaction au stress [14], notamment la
disparition des eets
permissifs et suppressifs
.
L’eet permissif sur l’action des catécholamines est
médié par diérents mécanismes: les glucocorticoïdes
augmentent la concentration de catécholamines cir-
culantes par induction de l’enzyme clé de la synthèse
d’adrénaline (phényléthanolamine N-méthyltransfé-
rase, PNMT) ou inhibition des enzymes responsables
de sa dégradation (par ex. catéchol-O-méthyltrans-
férase, COMT) et par des actions directes sur les récep-
teurs adrénergiques (par ex. augmentation de la capa-
cité de liaison ou de l’anité) ou l’inuence de
mécanismes post-récepteurs [14]. La suppression de ces
eets permissifs entraîne une résistance aux catécho-
lamines et ainsi une hypotension. Cette dernière est
renforcée par une déplétion hydrosodée (en raison
d’un décit en minéralocorticoïdes, de vomissements
ou de diarrhées).
L’eet suppressif des glucocorticoïdes empêche une ré-
ponse immunitaire excessive, par ex. par inhibition de
la sécrétion et de l’action de cytokines inammatoires
(TNF-α, IL-1 et IL-6) et antagonisme de la résistance aux
glucocorticoïdes médiée par les cytokines au niveau
du récepteur [15]. Outre la première manifestation
d’une insusance surrénalienne, les infections (avant
tout gastro-intestinales), les opérations, le stress émo-
tionnel ou la non-prise/non-augmentation du traite-
ment par glucocorticoïdes constituent les déclencheurs
d’une crise addisonienne les plus fréquents [16,17].
Causes (tab. 1)
Insusance surrénalienne primaire
A l’heure actuelle, la
surrénalite auto-immune
constitue
la cause la plus fréquente de l’
insusance surrénalienne
primaire
en Europe [18]. Avec une incidence de 4,4–6/
1 million d’habitants/an, il s’agit d’une maladie rare,
qui est toutefois diagnostiquée de plus en plus fré-
quemment [19]. La surrénalite auto-immune constitue
une maladie auto-immune «classique». Des méca-
nismes immunitaires cellulaires provoquent une des-
truction de l’ensemble de la glande surrénale. La mala-
La crise addisonienne illustre remarquablement
l’absence d’actions majeures des glucocorticoïdes
en cas de réaction au stress.
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(46):993–1003

ARTICLE DE REVUE MIG 996
die survient de manière isolée ou dans le cadre d’un
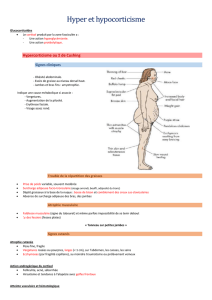
syndrome polyglandulaire auto-immun [20] (g.2a et
tab.1).
Dans les pays en développement, la tuberculose surré-
nalienne (g.2b) reste toujours la principale cause d’un
hypocorticisme primaire [21] et elle doit être considé-
rée en tant que diagnostic diérentiel chez les patients
originaires de ces pays.
La cause monogénique la plus fréquente d’insusance
surrénalienne primaire est le
syndrome adréno-génital
Tableau 1: Causes de l’insuffisance surrénalienne.
Cause Remarques
Insuffisance surrénalienne primaire
Surrénalite auto-immune (maladie d’Addison)
– Isolée (30–40%) Associée à HLA-DR3/-DR4
– Syndrome polyglandulaire auto-immun de type 1 (5–10%) Transmission autosomique récessive; mutations du gène AIRE (gène de la régulation auto-
immune); maladies associées: hypoparathyroïdie, candidose mucocutanée chronique, in-
suffisance ovarienne précoce, diabète sucré de type 1, dystrophies des tissus ectodermiques
(ongles, émail des dents), alopécie areata, hépatite auto-immune, anémie pernicieuse
– Syndrome polyglandulaire auto-immun de type 2 (60%) Associé à HLA-DR3/-DR4; maladies associées: hypo-/hyperthyroïdie, insuffisance ovarienne
précoce, diabète sucré de type 1, anémie pernicieuse, maladie cœliaque, vitiligo, alopécie
areata
Infectieuse
– Tuberculose surrénalienne Cause principale d’une insuffisance primaire dans les pays en développement
– Associée au VIH
– Infections fongiques
(par ex. histoplasmose, cryptococcose)
Avant tout chez les patients immunodéprimés
Maladies génétiques
– Syndrome adréno-génital Transmission autosomique récessive, le plus souvent, déficit en 21-hydroxylase
– Adrénoleucodystrophie Transmission liée à l’X
Hémorragie surrénalienne bilatérale/
infarctus surrénalien
Thrombopénie, syndrome de Waterhouse-Friderichsen, anticoagulation orale, syndrome
desantiphospholipides
Infiltration surrénalienne Métastases (par ex. carcinome bronchique, mammaire ou colique), amylose, sarcoïdose
Antécédent de surrénalectomie bilatérale
Insuffisance surrénalienne centrale
Glucocorticoïdes exogènes Principale cause d’une insuffisance centrale (anamnèse!)
– Antécédent de traitement d’un syndrome
de Cushing hypophysaire ou surrénalien
Tumeurs hypothalamo-hypophysaires Défaillance d’autres fonctions hypophysaires (hypopituitarisme), évtl. signe de croissance
tumorale locale (dégradations du champ visuel)
– Adénome hypophysaire Cause la plus fréquente
– Craniopharyngiome
– Méningiome
– Métastases (rares) Rares; le plus souvent, carcinome bronchique, mammaire ou colique
Hypophysite
– Lymphocytaire Isolée, en cas de syndrome polyglandulaire auto-immun, rare
– Traitement par inhibiteurs de point
de contrôle immunitaire
Anticorps anti-CTLA-4 (par ex. ipilimumab, jusqu’à 15%), anticorps anti-PD-1 / anti-PD-L1
(parex. pembrolizumab, jusqu’à 6%)
Apoplexie hypophysaire Adénome préexistant, traitement médicamenteux (par ex. analogues de la GnRH)
– Syndrome de Sheehan Survenue en péripartum (hypotension et perte de sang importante)
Maladies infiltratives Sarcoïdose, histiocytose langerhansienne, hémochromatose,
granulomatose de Wegener, tuberculose
Traumatisme cranio-cérébral
Antécédent de radiothérapie Latence possible de plusieurs années avant la survenue
Causes monogénétiques rares Chute isolée d’ACTH ou chute combinée de plusieurs hormones hypophysaires
Insuffisance surrénalienne d’origine médicamenteuse
Glucocorticoïdes (voir ci-dessus)
Inhibiteurs de point de contrôle immunitaire (voir ci-dessus)
Inducteurs du CYP3A4: ↑ dégradation du cortisol Mitotane, rifampicine, phénobarbital, phénytoïne, carbamazépine,
oxcarbazépine, topiramate
Inhibiteurs de la synthèse des hormones stéroïdes
surrénaliennes: ↓ production de cortisol
Mitotane, kétoconazole, fluconazole, itraconazole, métyrapone, étomidate
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(46):993–1003

ARTICLE DE REVUE MIG 997
(SAG) à transmission autosomique récessive, qui est
dans la plupart des cas causé par un décit en 21-hy-
droxylase.
L’
adrénoleucodystrophie
est une aection liée à l’X rare
caractérisée par un défaut au niveau de la biosynthèse
des stéroïdes, avec une accumulation d’acides gras à
très longue chaîne («very long chain fatty acids»,
VLCFA) principalement dans la glande surrénale et le
système nerveux central (SNC). Elle touche essentielle-
ment les hommes, les femmes étant des conductrices
de la maladie. Le plus souvent, des symptômes de l’at-
teinte du SNC (troubles comportementaux, décits
neurocognitifs allant jusqu’à l’évolution démentielle)
se manifestent déjà à un âge jeune.
Insusance surrénalienne centrale
Le traitement par
glucocorticoïdes exogènes
constitue
la cause la plus fréquente d’insusance surrénalienne
centrale [22]. Même un traitement oral de courte durée
entraîne déjà une insusance surrénalienne, généra-
lement passagère, chez plus de 40% des patients [23].
Outre les caractéristiques inhérentes à la substance,
telles que la demi-vie ou l’activité glucocorticoïde, ce
sont avant tout la durée et la dose cumulative qui sont
déterminantes pour le développement d’une insu-
sance surrénalienne iatrogène [24]. Tous ces facteurs
sont toutefois uniquement corrélés de manière impré-
cise avec le risque de développer eectivement un
hypocorticisme [25]. Une insusance surrénalienne
Figure 2: Observations en cas d’insuffisance surrénalienne primaire et centrale.
A: Patient avec syndrome polyglandulaire auto-immun de type 2: hyperpigmentation cutanée et vitiligo typiques.
B: Tuberculose surrénalienne: glandes surrénales calcifiées des deux côtés visibles à la radiographie abdominale.
C: Hyperpigmentation de la muqueuse buccale en cas de maladie d’Addison.
D: Hypophysite associée à l’ipilimumab: hypertrophie sphérique de l’hypophyse avec épaississement du pédicule (flèche).
SWISS MEDICAL FORUM – FORUM MÉDICAL SUISSE 2016;16(46):993–1003
 6
7
8
9
10
11
12
13
6
7
8
9
10
11
12
13
1
/
13
100%