Télécharger l`article au format PDF

© L’Encéphale, Paris, 2008. Tous droits réservés.
L’Encéphale (2008) Supplément 6, S194–S200
journal homepage: www.em-consulte.com/produit/encep
La plasticité synaptique
La plasticité synaptique est un des mécanismes cellulaires
qui expliquerait les phénomènes d’apprentissage et
mémorisation. Sa déÀ nition remonte à 1949 période à
laquelle le psychologue canadien Donald O. Hebb, énonçait
que lorsqu’un neurone prend part de façon répétée à
l’activation d’un autre neurone, l’efÀ cacité des connexions
entre ces neurones est augmentée. Les premières évidences
expérimentales de cette plasticité synaptique remontent
aux travaux de Bliss et Lomo en 1973 [5] qui montrent
que des changements brefs et répétitifs de l’activité des
synapses excitatrices dans l’hippocampe provoquent une
augmentation persistante de l’efÀ cacité de la transmission
synaptique appelée potentialisation à long terme (LTP pour
l’appellation anglo-saxonne Long Term Potentiation).
La LTP est un phénomène inductible chez l’animal en plaçant
une électrode de stimulation au niveau présynaptique et une
électrode d’enregistrement au niveau post-synaptique. Ces
électrodes permettent l’enregistrement d’un potentiel post
synaptique dont la latence et l’amplitude sont caractéristiques
de la région ciblée. Une stimulation répétitive de quelques
secondes permet d’augmenter de façon durable l’amplitude
de la réponse synaptique traduisant une augmentation
d’efÀ cacité synaptique prolongée. L’augmentation d’efÀ cacité
synaptique peut durer plusieurs jours, voire des semaines.
Ces phénomènes de LTP ont été reproduits dans différents
Effets des antipsychotiques sur la neuroplasticité :
données animales
TM. Jay(a, b)
(a) INSERM, Physiopathologie des Maladies psychiatriques, U894, Centre de Psychiatrie et Neurosciences, 75014 Paris
(b) Université Paris Descartes, Faculté de Médecine Paris
Introduction
La plasticité est une propriété fondamentale du système
nerveux nécessaire aux fonctions cérébrales normales.
La neuroplasticité fait référence à la capacité du système
nerveux à s’adapter aux changements environnementaux.
Elle inclut différents types de plasticité :
synaptique permettant le développement de connexions
entre les systèmes neuronaux et le remodelage des contacts
synaptiques en terme d’efÀ cacité et de densité ;
structurale au niveau des épines dendritiques, reliée à la
plasticité synaptique ;
la neurogenèse ou développement de nouveaux neurones.
Ces dernières années ont vu une révolution dans nos
concepts sur le fonctionnement du cerveau et sa plasticité,
y compris chez l’adulte. Il est maintenant accepté que le
cerveau forme et élimine à grande vitesse des synapses.
La capacité du cerveau adulte à augmenter les connexions
en réponse à un stimulus est importante, allant jusqu’à
une augmentation de la densité synaptique de 36 % en
24 heures. La zone sous-ventriculaire, située sur les parois
des ventricules latéraux, et la zone sous-granulaire dans
le gyrus denté de l’hippocampe produisent en permanence
de nouveaux neurones. Les cellules souches deviennent
des cellules précurseurs puis des neuroblastes et enÀ n des
neurones.
•
•
•
* Auteur correspondant.
E-mail : [email protected]
L’auteur n’a pas signalé de conÁ its d’intérêts.

Effets des antipsychotiques sur la neuroplasticité : données animales S195
circuits cérébraux et chez de nombreuses espèces animales y
compris l’homme. Chez ce dernier, une augmentation durable
(plusieurs heures) de l’efÀ cacité des synapses au niveau du
gyrus denté de l’hippocampe a récemment été reproduite sur
des coupes de lobe temporal de patients épileptiques [4].
La LTP est principalement limitée aux synapses
excitatrices et le transmetteur excitateur est le glutamate.
Lors d’une activation normale de la synapse, le glutamate
n’ouvre que des canaux non-NMDA (AMPA). Les récepteurs
NMDA qui permettent l’entrée de calcium (Ca2+) dans la
cellule sont inactifs au repos car ils sont bloqués par les
ions magnésium (Mg2+). La stimulation tétanique (induction
LTP) provoque une forte dépolarisation de la membrane via
l’entrée d’ions sodium (Na+) par les récepteurs AMPA qui
ouvrent les canaux NMDA (éjection de l’ion Mg2+) et permet
l’entrée de Ca2+ dans le neurone. L’entrée massive d’ions
Ca2+ est le facteur principal déclenchant la potentialisation
synaptique. Cet afÁ ux calcique va provoquer l’activation de
plusieurs protéines kinases dont la PKC, TK, CaMKIIŞ, PKA,
MAPK… L’activation de ces nombreuses kinases présentes au
niveau synaptique va permettre d’activer plusieurs voies de
signalisation cellulaire qui aboutiront à la potentialisation
de la synapse. La phosphorylation des récepteurs AMPA va
augmenter leur conductance aux ions Na+ et conjointement,
provoquer une augmentation du nombre de récepteurs
AMPA insérés dans la membrane postsynaptique. Ainsi, lors
de la prochaine stimulation nerveuse, il y aura facilitation
de la dépolarisation postsynaptique par augmentation
globale de l’efÀ cacité des récepteurs AMPA. L’efÀ cacité de
la synapse sera donc augmentée (Fig. 1). Conjointement et
via l’activation de facteurs de transcription, l’expression de
gènes et une synthèse de protéines de novo détermineront
le maintien ou non de la LTP.
La LTD (pour l’appellation anglo-saxonne Long Term
Depression) représente une diminution de l’efÀ cacité de
la transmission excitatrice glutamatergique. Phénomène en
miroir de la LTP, la LTD repose également sur une entrée
d’ions calcium via les récepteurs NMDA, mais en quantité
nettement moindre. À l’inverse de la LTP, l’expression de la
LTD se traduit par une diminution du nombre de récepteurs
AMPA au niveau de la synapse.
L’étude des mécanismes de la LTP et de la LTD a mis
en évidence une dynamique temporelle d’un ensemble de
mécanismes biochimiques et moléculaires qui permettent
la réorganisation des connexions synaptiques, la formation
de nouveaux réseaux neuronaux. Ces changements de
la force synaptique constituent les mécanismes les plus
vraisemblables de l’apprentissage et de la mémoire. Il existe
certes un faisceau convergent d’arguments en faveur d’un
lien éventuel entre LTP et mémoire, mais aucune preuve
n’est établie de manière certaine. Toutes les conclusions
ne convergent pas : par exemple, un apprentissage est
possible chez des souris knock-out dépourvues de capacité
de LTP.
De très nombreuses études reposent sur des approches
d’électrophysiologie sur tranches tissulaires menées ex vivo.
On ne sait pas si ces phénomènes identiÀ és et caractérisés
existent in vivo.
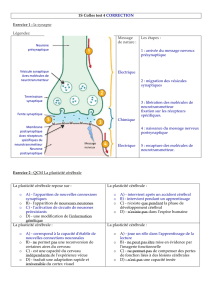
Figure 1 Mécanismes moléculaires de la LTP. Le transmetteur est le glutamate. Dans des conditions d’activation normale
des synapses (A), le glutamate n’ouvre que des canaux non-NMDA (AMPA), les récepteurs NMDA sont bloqués par l’ion Mg2+.
Lors de l’activation répétitive ou tétanique (induction) qui permet l’induction de la LTP (B), l’entrée d’ions sodium (Na+) par
les récepteurs AMPA engendre une dépolarisation sufÀ sante pour provoquer le déblocage des récepteurs NMDA par l’éjection
de l’ion Mg2+ et permettre l’entrée d’ions Na+ et Ca2+ dans le neurone. L’entrée d’ions Ca2+ déclencher l’activation de
cascades intracellulaires (PKC, TK, CaMK) qui auront pour conséquence une phosphorylation des récepteurs AMPA et une
incorporation de récepteurs AMPA supplémentaires dans la membrane post-synaptique (C).
CaM : Ca**-calmoduline
CaMK : Ca**-calmoduline kinase
FT : Facteurs de transcription
PKC : Protéinkinase C
TK : Tyrosine kynase
Terminaison
axonique
Transmission
synaptique normale
Induction
de LTP
Après
LTP
Épine
dendritique
Glu
Mg2+ Na+
NMDA AMPA
Pool latent de
R. AMPA
CaM
PKC TK CaMK
Terminaison
axonique
Épine
dendritique
Mg2+ Ca2+ Na+
NMDA AMPA
CaM
PKC TK
FT
CaMK
Terminaison
axonique
Épine
dendritique
Mg2+ Na+
NMDA AMPA
Pool latent de
R. AMPA
CaM
PKC TK CaMK
Na+
Ca2+
ABC

TM. JayS196
Effets des APA sur les capacités cognitives
et la plasticité synaptique
Les antipsychotiques atypiques, par rapport aux
neuroleptiques classiques, permettent l’amélioration de la
symptomatologie positive et des performances cognitives
[14, 17]. Lahti et coll. [13] montrent que chez des patients
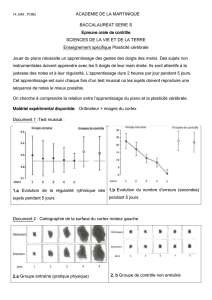
Figure 2 Potentialisation à long terme (LTP) induite au niveau du circuit hippocampe-cortex préfrontal chez les rats
contrôles, exposés à un stress aigu et chronique. A : Coupe schématique du cortex préfrontal montrant l’électrode
d’enregistrement (Rec) placée dans les cellules pyramidales du cortex préfrontal médian aÀ n d’enregistrer les potentiels
de champ évoqués par l’électrode de stimulation (« tetanized input ») placées dans l’hippocampe ventral (CA1/
subiculum) d’où partent les À bres en direction du cortex préfrontal. Les potentiels de champ enregistrés avant et après
l’application de la stimulation tétanique sont montrés en 1 (contrôles) et 2 (stressés) : L’amplitude ces potentiels de
champ est augmentée de façon durable (LTP) après l’application d’une stimulation tétanique. La LTP est bloquée après
exposition à un stress aigu (B) et chronique (C). Adaptée d’après [6, 20].
Rec.
Cortex préfrontal
Hippocampe
CA1/sub
Tetanized
input
A
60
80
100
120
140
160
180
– 30 0 30 60 90 120
Time (min)
Stressés
Contrôles
SHF
Changes in PSP amplitude
(% of baseline)
Stress
aigu
B
80
100
120
140
160
180
Stress
chronique
Stressés
Contrôles
SHF
– 30 0 30 60 90 120
Time (min)
Change in PSP amplitude
(% baseline)
C
schizophrènes, la clozapine normalise le débit sanguin
cérébral dans le cortex cingulaire lors d’une tâche cognitive
et rétablit une performance normale chez les patients
traités. Or, selon les données animales les performances
cognitives dépendent du système glutamatergique et des
récepteurs NMDA au niveau de l’hippocampe et du cortex
préfrontal.

Effets des antipsychotiques sur la neuroplasticité : données animales S197
Peu d’études se sont intéressées à l’effet des
antipsychotiques atypiques sur la LTP contrairement à celui
des antidépresseurs. Les premières études ont été menées
par l’équipe de Kubota et coll [10, 11]. Ces auteurs ont tout
d’abord rapporté un blocage de la LTP par l’halopéridol et
la carbamazépine. Ils ont ensuite mis en évidence que la
clozapine, administrée pendant 3 semaines à la dose de
20 mg/kg chez le lapin, potentialise la réponse synaptique
avant la LTP hippocampique et non l’induction de la LTP.
Cette potentialisation dépend des récepteurs NMDA et induit
une libération de dopamine au niveau de l’hippocampe.
La rispéridone administrée de manière chronique à
la dose de 10 et 20 mg/kg chez le lapin n’a pas d’effet
inducteur de la LTP hippocampique [12].
Effets des APA sur les effets délétères
du stress sur la plasticité synaptique
Le stress bloque l’efÀ cacité des synapses du système
hippocampe – cortex préfrontal. Lorsqu’on applique un stress
aigu comportemental à des animaux, on met en évidence
une augmentation des corticostérones plasmatiques et
un blocage de la LTP, témoin du blocage de la plasticité
synaptique par le stress [20].
Le stress chronique entraîne aussi un blocage de la
LTP chez l’animal, ainsi qu’un déÀ cit de performances
cognitives comme la mémoire de travail et la Á exibilité
comportementale [6]. La Á exibilité comportementale fait
référence à la capacité de l’animal à s’adapter à un nouvel
environnement.
Les relations entre stress, fonctions cognitives et
circuit hippocampe-cortex prefrontal et le rétablissement
d’une fonction préfrontale normale sous clozapine [13]
nous ont conduit à investiguer les effets de la clozapine
sur les perturbations de plasticité synaptique hippocampo-
préfrontale observées chez l’animal. Nous avons montré
que la clozapine en traitement aigu diminue les effets
négatifs du stress sur la plasticité prefrontale [19]. Ces
résultats suggèrent que, sous clozapine, les fonctions
cognitives peuvent être maintenues après stress ou que
la clozapine est capable de rétablir la balance au niveau
des synapses hippocampo-préfrontales. Faisant référence
aux travaux de Sebban et coll. [21], qui avaient montré
des effets (dose dépendants) de la clozapine sur l’activité
theta frontale chez des rats vigils, nous avons utilisé une
dose bien inférieure à ce qui est classiquement utilisé.
En effet, l’activité theta au niveau frontal joue un rôle
majeur dans la mémoire de travail chez plusieurs espèces
incluant l’homme et cette activité est très importante dans
l’expression de la plasticité synaptique.
Plasticité structurale
La plasticité structurale est directement reliée à la plasticité
synaptique. Sur le plan structural, les changements
dynamiques sont localisés au niveau des épines dendritiques.
Ces dernières sont le site majeur de l’excitation post-
synaptique. La plupart des cellules pyramidales dans le
cortex cérébral ont des milliers d’épines le long de l’arbre
dendritique. Grâce aux nouvelles techniques d’imagerie
(microscopie confocale), on sait que les épines dendritiques
sont en perpétuel changement. Récemment on a pu appliquer
une stimulation répétitive des récepteurs NMDA au niveau
des épines dendritiques des cellules CA1 de l’hippocampe et
montrer que cette procédure de stimulation augmente de 28 %
le diamètre des épines. Cette augmentation de taille dépend
de la taille dendritique initiale. Chez le sujet souffrant de
schizophrénie, on observe une diminution du volume du cortex
préfrontal, une réduction de la densité et de la longueur des
épines dendritiques ainsi qu’une diminution des terminaisons
présynaptiques et de la taille du soma neuronal.
Il est clairement établi que ces processus dynamiques sont
régulés dans des conditions normales et pathologiques.
Effets des APA sur cette plasticité
structurale
Une première étude menée par l’équipe de Critchlow et
coll. montre que la spinophiline (phosphatase spéciÀ que
des épines dendritiques) est augmentée de 70 % dans des
neurones hippocampiques cultivés in vitro en présence
de clozapine (1 μM). Lorsque les auteurs examinent
les dendrites de ces cellules en microscopie confocale,
ils détectent une augmentation plus importante de la
densité des épines dendritiques en présence de clozapine
comparativement à l’halopéridol [7].
Plus récemment, Wang et Deutch se sont intéressés
au devenir des terminaisons glutamatergiques dans le
cortex préfrontal après un déÀ cit dopaminergique [24].
Dans un premier temps, les auteurs ont étudié l’effet
d’une lésion par la 6 hydroxy-dopamine sur les épines
dendritiques in vivo. Puis ils se sont intéressés à l’impact
des antipsychotiques (halopéridol ou olanzapine) sur les
changements structuraux. Après 3 semaines, ils observent
une diminution de la longueur et de la complexité de
l’arbre dendritique des cellules pyramidales du cortex
préfrontal chez les rats lésés traités avec une solution
saline. Les rats lésés traités par halopéridol ne présentent
pas un net changement au niveau de la longueur et des
branchements dendritiques au niveau basal. En revanche,
il existe une forte augmentation des épines dendritiques
chez les animaux traités par olanzapine.
Neurogenèse
La neurogenèse, aboutissant à la production de nouveaux
neurones, est restreinte à deux zones cérébrales : zones sous
granulaire au niveau du gyrus denté et sous ventriculaire.
Sa découverte chez l’adulte a modiÀ é la compréhension
de la plasticité cérébrale jusqu’alors considérée comme
étant uniquement d’ordre synaptique.
De nouveaux neurones naissent et meurent quotidienne-
ment dans le gyrus denté de l’hippocampe adulte chez tous
les mammifères, y compris chez l’homme. La neurogenèse
dans le cerveau adulte est un phénomène de différenciation
par lequel les cellules souches de l’hippocampe, se divisent :

TM. JayS198
une cellule demeure cellule souche, l’autre se transforme
en neurone. Cette forme de plasticité neuronale est régu-
lée par de nombreux facteurs physiologiques dont certains
paraissent impliqués dans les troubles de l’humeur.
Le stress chronique via une augmentation du cortisol
plasmatique réduit le nombre de nouveaux neurones
générés dans l’hippocampe alors que l’exercice physique,
les antidépresseurs et le lithium l’augmentent [8].
La technique employée pour détecter la production
de nouveaux neurones dans le cerveau est relativement
simple. La plupart des études ont utilisé un analogue de
la thymidine, la bromodésoxyuridine (BrdU). Lorsque cette
molécule est administrée à un animal, elle s’incorpore dans
le nouveau brin d’ADN qu’une cellule synthétise peu avant
la mitose. Il est alors possible, a posteriori, de retrouver la
trace des cellules qui ont incorporé le marqueur de synthèse
grâce à des anticorps spéciÀ ques anti-BrdU. En combinant
des marqueurs neuronaux spéciÀ ques du BrdU, on parvient
à prouver que les cellules nouvellement générées sont
effectivement des neurones et non des cellules gliales.
Effets des APA sur la neurogenèse.
Implication des facteurs trophiques
Un travail mené par Newton et Duman en 2007 [18], résume les
données à propos des effets des antipsychotiques atypiques
sur la neurogenèse (Tableau 1). L’olanzapine apparaît
comme la molécule augmentant le plus la neurogenèse dans
les zones sous granulaire et sous ventriculaire mais aussi
dans le cortex préfrontal. Cependant la neurogenèse dans
le cortex préfrontal n’est pas observée dans des conditions
physiologiques normales. L’olanzapine à long terme induit
une augmentation de 56 % de la prolifération cellulaire non
neuronale dans le cortex préfrontal.
Ces données rejoignent les résultats de Selemon et al. chez
le singe : après l’administration d’antipsychotiques atypiques
pendant 6 mois, on observe une prolifération gliale [22].
Chez le rat, l’administration de quétiapine à la dose de
10 mg/kg pendant 7 ou 21 jours normalise la neurogenèse
hippocampique après exposition au stress [16, 25].
Si les antipsychotiques atypiques ont un rôle dans
la neurogenèse, est-ce par un effet sur les facteurs
trophiques ? Les études ont essentiellement porté sur Brain
Derived Neurotrophic Factor (BDNF), Nerve Growth Factor
(NGF), et VGF. Les résultats publiés sont contradictoires.
La rispéridone et l’halopéridol semblent diminuer
l’expression du BDNF dans l’hippocampe, le cortex préfrontal
et orbital [1] contrairement à l’olanzapine (2,7 mg/kg) et
à la clozapine (10 mg/kg) au niveau hippocampique [3].
L’olanzapine augmenterait le NGF, la phosphorylation des
voies de signalisation (Akt, ERK) impliquées dans la production
de BDNF [15]. La clozapine (25 mg/kg), l’olanzapine (2,5 mg/
kg, 5 mg/kg, 10 mg/kg), et l’aripiprazole (2 mg/kg, 10 mg/
kg, 20 mg/kg) n’auraient pas d’effets sur les taux protéiques
de BDNF et de NGF hippocampiques [23].
On peut donc s’interroger sur la nature des différences
entre APA et neuroleptiques classiques. Tout d’abord à
propos de l’action sur le BDNF : les antagonistes 5HT2A
Tableau 1 Induction de la neurogenèse et de la prolifération cellulaire par les antipsychotiques atypiques (d’après 18)
Treatment Brain regionaEffect Reference
Drug Dos (mg/kg) No. days
Atypical
Olanzapine 2 21 SGZ Increased 52
Olanzapine 10 21 SGZ Increased 53
Clozapine 0.5 – 20 28 SGZ Increased 54
Risperidone 0.5 21 SGZ No effect 55
Olanzapine 2 21 SGZ No effect 55
Olanzapine 2 21 SVZ No effect 52
Risperidone 0.5 21 SVZ Increased 55
Olanzapine 0 21 SVZ Increased 55
Olanzapine 2 21 PFC Increased 52
Olanzapine 10 21 PFC Increased 53
Olanzapine 10 21 Striatum Increased 53
Olanzapine 2 21 Striatum Increasedb52
Olanzapine 2 21 SVZ, PFC Increased 56
Olanzapine 2 21 SGZ No effect 56
Risperidone 0.5 21 SVZ, SGZ No effect 56
a. SGZ and SVZ refer to neurogenesis : PFC and striatum refer to proliferation of non-neuronal cells.
b. Trend for increased cell proliferation.
OB = olfactory bulb ; PFC = prefrontal cortex ; SGZ = subgranular zone ; SVZ = subventricular zone.
 6
7
6
7
1
/
7
100%