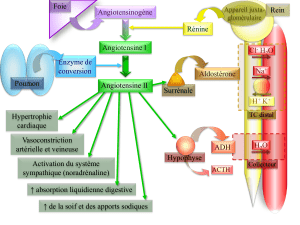
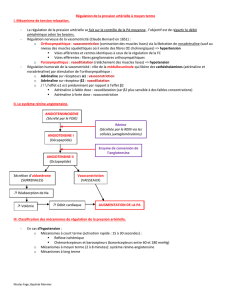
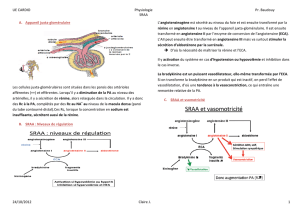
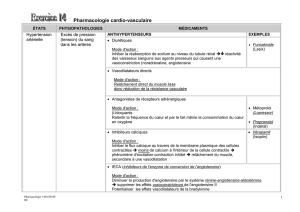
le systeme renine-angiotensine- aldosterone chez le chien

ANNEE 2007 THESE : 2007-TOU3-4038
…
LE SYSTEME RENINE-ANGIOTENSINE-
ALDOSTERONE CHEZ LE CHIEN SAIN ET LE
CHIEN INSUFFISANT CARDIAQUE
_________________
THESE
pour obtenir le grade de
DOCTEUR VETERINAIRE
DIPLOME D’ETAT
présentée et soutenue publiquement en Avril 2007
devant l’Université Paul-Sabatier de Toulouse
par
Blandine, Danièle, Jeanine MOREL
Née, le 30 décembre 1981 à SAINT-ETIENNE (42)
___________
Directeur de thèse : Mme le Docteur Armelle DIQUELOU
___________
JURY
PRESIDENT :
Mme Elisabeth Arlet-Suau
ASSESSEUR :
Mme Armelle DIQUELOU
M. Patrick VERWAERDE
Professeur à l’Université Paul-Sabatier de TOULOUSE
Maître de Conférences à l’Ecole Nationale Vétérinaire de TOULOUSE
Maître de Conférences à l’Ecole Nationale Vétérinaire de TOULOUSE

2

3
REMERCIEMENTS
A notre président de jury,
A Madame le professeur Elisabeth Arlet-Suau
Professeur des Universités
Praticien hospitalier
Médecine interne
Qui nous a fait l’honneur d’accepter
la présidence de notre jury de thèse.
Hommages respectueux.

4
A notre jury de thèse,
A Madame le Docteur Armelle Diquelou
Maître de Conférences de l’Ecole Nationale Vétérinaire de Toulouse
Pathologie médicale des équidés et carnivores,
Qui a bien voulu accepter de diriger cette thèse,
Merci pour son soutien, sa disponibilité et sa gentillesse,
Qu’elle trouve ici l’expression de mes remerciements.
et de mon respect le plus sincère.
A Monsieur le Docteur Patrick Verwaerde
Maître de Conférences de l’Ecole Nationale Vétérinaire de Toulouse
Anesthésie, réanimation,
Qui nous a fait l’honneur de participer à notre jury de thèse,
Sincères remerciements.
A mes parents,
Pour votre présence et votre soutien pendant mes études.

5
Trouvez dans ce travail tout mon amour et ma reconnaissance.
A ma mamie,
Pour tes encouragements, tes conseils et ton amour. Merci de m’avoir appris à prendre
confiance en moi.
Trouve dans ce travail tout mon amour.
A ma sœur, Typhanie,
A nos moments de complicité. Je te souhaite bonheur et réussite dans ta nouvelle vie. Que tes
projets se concrétisent.
A Thomas,
Le nouveau venu dans la famille… Bon courage. Je te souhaite plein de joie dans ta vie.
A Gaëlle,
Pour avoir été toujours là pendant ces nombreuses années malgré la distance. A nos rêves
d’adolescentes. En espérant que ces dix années d’amitié ne soient que les premières d’une
longue série.
A toutes les poufs : Alexandra, Aude, Marion, Françoise, Audrey, Béatrice,
Karine,Vanessa et Sophie,
En souvenir de tous ces moments de complicité et de bonheur inoubliables passés à l’école,
les soirées, les apéros improvisés du vendredi soir, les week-end et les nuits de gardes… En
espérant que les années ne nous éloignent pas.
A Magali,
En souvenir de notre complicité pendant cette dernière année de prépa qui m’a paru moins
dure à tes côtés. Notre amitié n’en est que plus forte.
A Ingrid,
A une amitié naissante. Je te souhaite tout le bonheur que tu mérites.
Aux vétérinaires qui m’ont donné la chance de commencer et plus particulièrement au
Docteur Pernoud, au Docteur Reboul et leurs équipes.
A tous les animaux qui m’ont accompagné vers le chemin de l’école. A mon p’tit Lou,
Sirocco, Bambou, Gari, Patachon, Grisou, Astro, Fafoune et à tous ceux à venir.
Merci à tous ceux que je n’ai pas cité mais que je porte dans mon cœur.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
1
/
165
100%