1

2
Introduction
Il existe dans la cellule 2 grands types de réactions biochimiques :
- les réactions de dégradation de la matière organique (catabolisme);
- les réactions de synthèse de la matière organique (anabolisme).
Ces réactions métaboliques se déroulent dans la cellule en présence d’un catalyseur biologique
(biocatalyseur) encore appelé enzyme
.
Les enzymes sont donc des composés biologiques de
nature protéique, doués d’activité catalytique et produites par la cellule vivante. Leurs
anciennes appellations sont les ferments ou les diastases. On écrira une réaction enzymatique de
la manière suivante:
Une enzyme fixe un substrat pour le transformer en produit. Ainsi, l'enzyme possède un
site actif formé de l'ensemble des acides aminés qui entrent en contact avec le substrat. Ce
site actif est formé de deux sites: le site de fixation du substrat et le site catalytique qui va agir
sur le substrat et lui faire subir la réaction chimique. Les acides aminés qui constituent le site
catalytique sont en général la sérine, la cystéine, l'histidine, la tyrosine et la lysine. Le site de
fixation du substrat et le site catalytique peuvent être confondus ou distincts. Certaines enzymes
sont en plus capables d'assurer la régulation des chaînes métaboliques. Cette faculté est due à
la présence d'un site régulateur (en plus du site actif) qui réagit avec des modulateurs qui viennent
d'ailleurs (allostérie). C'est le cas des enzymes allostériques.
Le rôle des enzymes est d’accélérer les vitesses des réactions biochimiques dans la
cellule. Un catalyseur est une substance qui, sans éprouver de transformation visible, et à faible
dose, modifie la cinétique d’une réaction chimique. Ainsi, la catalase catalyse la réaction de
transformation de l’eau oxygénée en une eau simple et en oxygène moléculaire:
H
2
O
2
→H
2
O + ½ O
2
Avec une concentration de H2O2
=
1
MOL
/L, la vitesse de réaction est de 2,2.10
–13
mol/L/s sans catalyseur, de 5,4.10
–9
mol/L/s avec un catalyseur chimique (le platine colloïdal) et
de 4,5.10
–2
mol/L/s en présence de catalase.
Une enzyme donnée est spécifique d’une réaction, c’est-à-dire qu’elle catalyse toujours
la même transformation, se produisant sur les mêmes corps chimiques initiaux. Les protéines
enzymatiques sont synthétisées par des êtres vivants. Cette synthèse est déterminée génétiquement

3
: sa conservation dans le génome est favorisée par le besoin qu’éprouve cet être vivant de faire
cette réaction. Toutes les molécules qui entrent dans une réaction enzymatique et sont
définitivement modifiées sont appelées substrats. Le substrat est donc une molécule de nature
organique ou inorganique reconnue par le centre actif de l’enzyme et transformé par celui-
ci. La nouvelle molécule qui résulte de cette transformation est appelée produit. Toutes les
molécules ayant une liaison spécifique avec une protéine sont appelées ligands. Pour chaque
ligand, il existe au moins un site de fixation sur la protéine qui le reçoit. D’autres molécules non
protéiques peuvent aussi avoir une activité catalytique : les abzymes (Ac) et les ribozymes
(ARN). L’enzymologie est la science qui étudie les enzymes. Le spécialiste de cette science est
appelé enzymologiste. Le substantif « enzyme » est du genre féminin et masculin.

4
Caractéristiques générales des enzymes
I- Propriétés caractéristiques des enzymes
-Les enzymes respectent les lois générales de la catalyse:
•le catalyseur agit en quantité très faible et se retrouve intact à la fin de la réaction;
•le catalyseur ne modifie ni la nature de la réaction, ni son équilibre thermodynamique;
•il accélère la vitesse de la réaction.
-Les enzymes abaissent davantage l’énergie d’activation des réactions chimiques en augmentant la
probabilité des chocs efficaces. L’énergie d’activation ou l’énergie libre d’activation (ΔG
A
) est
l’énergie qui doit être absorbée par les réactifs pour que leurs liaisons soient brisées. Les
réactifs doivent atteindre un état de transition instable dans lequel les liaisons sont plus fragiles et
plus faciles à briser. Même une réaction exergonique où le ΔG est négatif, requiert l’absorption
d’énergie pour atteindre l’état de transition.
Les enzymes ont la capacité d’abaisser l’énergie d’activation pour des réactions spécifiques
pour que ces réactions puissent se faire à la température normale de la cellule.
-Les enzymes présentent une grande spécificité compte tenu de leur nature protéique ;
-Leur activité peut-être modulable en fonction de l’environnement : pH, T°, Substrat, composés
activateurs ou inhibiteurs.
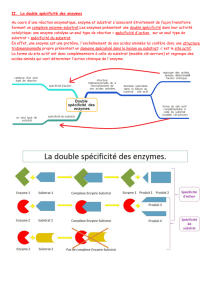
II- Spécificité de l’action enzymatique
La spécificité est l’une des caractéristiques principales des enzymes. Elle peut être circonscrite à
4 niveaux.

5
1- Stéréospécificité ou spécificité stéréochimique
Beaucoup d’enzymes sont capables de distinguer deux isomères optiques (séries L et
D) l’un de l’autre. Elles peuvent agir sur le composé de la série L et demeurer inactives sur le
composé de la série D ou vice versa. On dit que les enzymes sont adaptées stéréochimiquement
à leurs substrats. C’est le cas de la:
-D-aminoacide déshydrogénase du foie des mammifères qui catalyse la déshydrogénation
d’un grand nombre de D-aminoacides et est inactive sur les L-aminoacides:
-lactico-déshydrogénase du muscle qui peut catalyser la déshydrogénation de l’acide L(+)-
lactique en acide pyruvique mais non celle de l’acide D(-)lactique;
-l’arginase qui n’hydrolyse par contre que la L(+)arginine en ornithine et urée;
-d’un certain nombre d’enzymes font la distinction entre les conformations alpha et
beta. C’est le cas par exemple des alpha et beta-glucosidases, galactosidases, fructosidases etc ;
D’autres enzymes ont une stéréospécificité vis à vis de l’isomérie cis-trans.
2- Spécificité liée à la nature de la liaison
La nature de la liaison est la condition nécessaire pour que la substance puisse être
dégradée. Ainsi, dans le cas des hydrolases, on distingue selon le type de liaison, des osidases, des
estérases, des amidases, etc. Les estérases hydrolysent seulement les liaisons esters et non les
liaisons osidiques ou peptidiques. Parmi les estérases, on peut faire la distinction entre les
carboxy-estérases, spécifiques des liaisons esters où sont engagés des acides organiques, et les
phospho-estérases qui hydrolysent les esters phosphoriques. Parmi les enzymes protéolytiques
encore appelées protéases (enzymes hydrolysant les liaisons peptidiques) on distingue les exo-
peptidases qui exercent leur action à l’extrémité des chaînes et détachent l’amino-acide N- ou C-
terminal et les endopeptidases qui hydrolysent les liaisons peptidiques à l’intérieur des chaînes.
3- Spécificité de groupe
L’enzyme exigera que son substrat possède une liaison donnée et au moins une partie de la
molécule conforme à une structure définie (A ou B). C’est le cas de la trypsine, enzyme
protéolytique contenu dans le suc pancréatique qui hydrolyse les liaisons peptidiques des protéines
mais seulement celles auxquelles participent les groupements carboxyles de résidus de lysine ou
d’arginine. C’est le cas des a-glucosidases qui hydrolysent les liaisons osidiques entre l'a-glucose
et un alcool ou un ose. La nature de ces liaisons influencera la vitesse de la réaction .
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
1
/
128
100%