Growth Hormone Treatment - Turner Syndrome Society of Canada

À propos du
syndrome de Turner
Qu’est-ce que le syndrome de Turner?
Le syndrome de Turner (ST) est un état qui ne touche que
les filles et les femmes. Il est déterminé par une différence
dans la composition génétique de celles qui sont touchées.
La plupart des filles et des femmes ont deux chromosomes
X entiers (les chromosomes sexuels chez les femelles). Le
syndrome de Turner est causé par l’absence de la totalité
ou d’une partie du second chromosome X chez certaines
(ou l’ensemble) des cellules du corps. Les caractéristiques
les plus constantes du ST sont une petite taille et une
absence de développement ovarien.
Une forme synthétique de l’hormone de croissance humaine
(HCH) est disponible à des fins médicales. L’HCH est
parfois administrée aux filles ayant le ST pour les aider à
grandir. La thérapie HCH peut servir à traiter les cas de
déficience en hormone de croissance ou de petite taille.
À l’âge typique de la puberté, les filles atteintes du ST se
voient habituellement prescrire la thérapie de substitution
hormonale pour induire les modifications du développement
féminin normales. Cette thérapie est souvent soutenue
durant la majeure partie de l’âge adulte.
Qu’est-ce qui cause le syndrome de Turner?
Le syndrome de Turner survient lorsqu’un élément des
renseignements génétiques (une partie ou la totalité du
chromosome X) « échappe » au processus appelé méiose,
au cours duquel les cellules sexuelles se divisent pour
former le sperme chez les mâles et les ovules chez les
femelles. À ce jour, on présume que le ST est lié à des
facteurs environnementaux ou à tout autre facteur
généralement associé aux problèmes génétiques.
Combien courant est le ST?
Le syndrome de Turner touche environ 1 femelle sur 2 500
nées parmi la population.2 Par conséquent, on chiffre à plus
de 7 000 le nombre de personnes ayant le ST au Canada.
Y a-t-il différentes formes du syndrome de
Turner?
Les filles auxquelles il manque un chromosome X entier
ont le ST « classique » ou le caryotype 45,X. Un
caryotype correspond à tout l’ensemble des
chromosomes présents dans les cellules d’une
personne. Plus de la moitié (environ 54 %)1 des femmes
ayant le syndrome de Turner ont le caryotype 45,X. Les
autres n’ont en moins qu’une partie du second
chromosome X, éprouvent une réorganisation de la
structure du chromosome ou ont un caryotype en
mosaïque, ce qui renvoie à un chromosome X
manquant ou réorganisé chez certaines (mais non la
totalité) des cellules du corps. Il est important qu’une
personne avec le ST connaisse son caryotype étant
donné que différents caryotypes sont associés à
différents troubles de santé potentiels.
Quels signes indiquent la présence du ST?
Voici les raisons les plus courantes qui
motivent une étude à savoir si une fille ou une
femme a le ST, mais ce ne sont pas là les
seuls motifs :
Nourrissons : petite taille, mains et pieds
potelés, plis cutanés supplémentaires sur le
côté et à l’arrière du cou, et anomalies
cardiaques.
Jeunes enfants : petite taille par rapport à
leurs pairs (voir ci-dessous un diagramme de
croissance « normal » pour ce qui est de la
hauteur et du poids), ainsi que d’autres signes
tels que des infections d’oreille et des
problèmes d’audition à répétition.
Adolescentes : petite taille, et absence de
développement des tissus mammaires ou de
menstrues à l’âge prévu.
Adultes : menstrues irrégulières, problèmes
de fécondité, petite stature, ainsi que troubles
de l’audition, anomalies cardiaques et
problèmes de tension artérielle.
Nous joindre
Société canadienne du syndrome de
Turner
30, av. Cleary, Pièce 9
Ottawa (ON), CANADA K2A 4A1
Sans frais : 1-800-465-6744
Tél. : 613-321-2267
Téléc. : 613-321-2268
Courriel : [email protected]
Website: www.turnersyndrome.ca

Comment diagnostique-t-on le ST?
Les filles et les femmes reçoivent un diagnostic à diverses
étapes de leur vie, de l’étape prénatale à la vie adulte. L’âge
du diagnostic n’a cessé de diminuer avec une meilleure
sensibilisation au ST parmi la collectivité médicale.
Généralement, il y a quelque chose de « différent » à propos
de la fille ou de la femme, ce qui incite le parent ou le
médecin à enquêter sur la possibilité d’une présence de ST.
On pose un diagnostic de ST lorsqu’il y a chez la personne
tout un ensemble de chromosomes appelé caryotype.
L’analyse du caryotype montre si une des paires de
chromosomes X est manquante par rapport à l’ensemble de
chromosomes complet, ou s’il y a des différences de
structure dans les chromosomes X.
Avant la naissance, on pose un diagnostic de ST en
prélevant un échantillon de liquide amniotique ou tout autre
tissu fœtal pour étudier le caryotype fœtal. Une échographie
est également utilisée pour dépister les motifs souvent
constatés chez le ST, notamment une accumulation de
fluide autour du cou, et des anomalies rénales ou
cardiaques. Après la naissance, on confirme le diagnostic de
ST en prélevant un échantillon de sang ou d’autres tissus
pour obtenir un caryotype.
Pourquoi est-ce important de savoir si
quelqu’un a le syndrome de Turner?
Il y a divers défis de santé, de développement, d’intégration
sociale et d’apprentissage qui peuvent nuire aux filles et aux
femmes ayant le ST dans différentes mesures. Il est
important de diagnostiquer le ST le plus tôt possible pour
que le médecin puisse déterminer si la fille ou la femme a
des problèmes de santé qui nécessitent un traitement ou qui
doivent être surveillés de près. Plus tôt on détermine les
problèmes d’apprentissage et de santé, meilleures sont les
chances que la personne avec le ST puisse recevoir l’aide
dont elle a besoin pour réussir.
Le syndrome de Turner peut-il être traité?
Le ST n’est pas une maladie, mais un état génétique qui est
associé à diverses préoccupations en matière de santé. Le
ST n’est pas directement « traité » par un médecin, mais les
préoccupations de santé individuelles de chaque fille ou
femme atteinte du ST doivent être surveillées et réglées par
le spécialiste approprié. Par exemple, les filles et les
femmes avec des conditions cardiaques couramment
repérées chez les personnes atteintes du ST devraient faire
l’objet d’un suivi par un cardiologue. Les avancées
médicales permettent aux femmes atteintes du ST et
intéressées à fonder une famille de solliciter l’aide d’un
obstétricien qui se spécialise en fécondation in vitro. Une fille
avec le ST peut obtenir une hormone de croissance sous la
supervision d’un endocrinologue pédiatre.
Mars 2014 Un merci tout spécial à Mme Melissa Pasqua, à la Dre Stasia
Hadjyannakis, à la Dre Jill Hamilton, à la Dre Karine Khatchadourian, au Dr Munier
Nour et à Mme Brenda Fraser pour leur aide et leur appui lors de la création de cette
Infocapsule. Traduction Luc C. Carriére. La traduction, l'impression et la
distribution de cette infocapsule ont été appuyées par le Programme ‘Lilly
Giving’ de Eli Lilly Canada Inc.
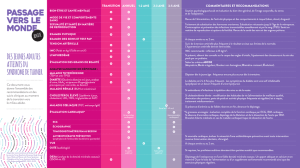
Quels problèmes de santé et d’intégration sociale pourraient
être liés au ST?
Chaque personne avec le ST est unique. Cependant, il y a
certaines caractéristiques, soit des problèmes potentiels ou des
attributs physiques, qui sont liées au ST. Veuillez noter que, sauf
pour les détails sous « Constatations conséquentes avec le ST »,
tous les autres aspects sont variables et ne sont pas recensés
chez chaque personne ayant le ST. Différents caryotypes
mèneront à différentes probabilités de ces constatations.
Constatations conséquentes avec le ST
- petite taille (hauteur moyenne de 142 cm)
- insuffisance ovarienne et l'infécondité
Aspects physiques (autres que la taille)
- large poitrine (55 %)3
- large et court cou avec excès de peau qui relie le cou à la
clavicule (« cou palmé ») (41 %)3
- lymphoedème ou accumulation de liquides (mains/pieds)
(36 %)3
Problèmes sociaux et comportementaux
- problèmes d’alimentation en bas âge et à l’âge adulte (70 %)4
- défis d’apprentissage visuel et spatial (70 %)2
- TDAH (24 %)5
- aptitudes sociales ardues
Problèmes de santé potentiels
- problèmes d’audition, y compris pertes auditives attribuables à
des infections chroniques ou récurrentes de l’oreille moyenne
(plus de 60 %)6
- anomalies des reins ou des voies urinaires (40 %)7
- anomalies cardiaques (30 %)8
- hypothyroïdie (23 %)9
- hypertension artérielle (plus de 25 %)10
- obésité
Problèmes de santé peu probables (moins de 20 %)
- problèmes oculaires tels que la ptose ou l’affaissement des
paupières (13 %)7 ou strabisme ou « œil paresseux » (18 %)7
- courbures de la colonne vertébrale, scoliose (10%)7
- diabète de types 1 et 2 (7 % de risque de l’un ou de l’autre)4
- ostéoporose et probabilité accrue de fractures (risque 10 fois
plus grand à comparer à la population d’ensemble)11
- cataractes (~ 8 %)12
- affections auto-immunes (maladie coéliaque, ostéoarthrite, etc.)
Références
1. Mazzanti et Cacciari, J Pediatr, nov. 1998; 133(5):688.
2. Sybert, V.P., McCauley, E., N Engl J Med, 16 sept. 2004;
351(12): 1227-38.
3. Charney, S., Smillie, A. (1983), The X’s and O’s of Turner’s
Syndrome, York University.
4. Chen, H., Faiaenbaum, D., Weiss, H. (1981), American
Journal of Medical Genetics, 8, 191-203.
5. Russel, H.F. et al., J. Pediatr. Psychol., oct. 2006; 31(9): 945.
6. Morimoto et al., J. Pediatr., nov. 2006; 149(5): 697.
7. Sybert, V.P., « Turner syndrome », Management of genetic
syndromes, 2001, New York: Wiley-Liss, 459-484.
8. Volkl et al., Clin Cardiol., fév. 2005; 28(2): 88-92.
9. Livadas, S. et al., Thyroid, 2005; 15: 1061-1066.
10. Bondy et al., J. Clin. Endocrinol. Metab., 2007; 92(1): 10-25.
11. Gravolt et al., J. Clin. Epidemiol., 1998; 51(2): 147.
12. Lessell, Forbes, Arch Opthalmol., 1966; 76(2): 211-213.
1
/
2
100%