FACULTE DE MEDECINE HENRI WAREMBOURG Mise en place d

UNIVERSITE DU DROIT ET DE LA SANTE - LILLE 2
FACULTE DE MEDECINE HENRI WAREMBOURG
Année : 2014
T H E S E P O U R L E D I P L O M E D ' E T A T
D E D O C T E U R E N M E D E C I NE
Mise en place d’un test fonctionnel pour évaluer les Variants de
Signification Inconnue des gènes MLH1 et MSH2 responsables du
syndrome de Lynch
Présentée et soutenue publiquement le vendredi 20 juin à 14 heures
au Pôle Recherche
Par Catherine KAZMIERCZAK-VERMAUT
_______________
JURY
Président :
Madame le Professeur Sylvie MANOUVRIER
Assesseurs :
Madame le Professeur Nicole PORCHET
Madame le Docteur Marie-Pierre BUISINE
Madame le Docteur Sophie LEJEUNE
Monsieur le Docteur Stéphane CATTAN
Directeur de Thèse :
Madame le Docteur Marie-Pierre BUISINE
_______________

Avertissement
La Faculté n'entend donner aucune approbation aux opinions émises dans les
thèses : celles-ci sont propres à leurs auteurs.

Liste des abréviations
CCR : cancer colorectal
IDL : Insertion/Deletion Loop
IHC : immunohistochimie
INCa : Institut National du Cancer
kDa : kilodalton
MMR : Mismatch Repair
MSI : Microsatellite Instability
MSS : Microsatellite Stability
NGS : Next Generation Sequencing
Pb : paire de base
UMD : Universal Mutation Database
VSI : Variant de Signification biologique et clinique Inconnue
WB : Western Blot

Table des matières
Introduction ................................................................................................................... 1
I. Le syndrome de Lynch : Physiopathologie, présentation clinique et prise en
charge ...................................................................................................................... 1
A. Généralités .................................................................................................................... 1
B. Physiopathologie ........................................................................................................... 2
1. Le système MMR ......................................................................................... 2
2. Mécanisme d’inactivation du système MMR dans le syndrome de Lynch .. 5
3. L’instabilité microsatellitaire : marqueur de l’inactivation du système
MMR ............................................................................................................ 6
C. Présentation clinique ..................................................................................................... 7
D. Prise en charge ............................................................................................................ 14
1. Recommandations de l’INCa .................................................................. 14
2. Suggestions du consortium européen ...................................................... 16
II. Stratégie diagnostique .......................................................................................... 17
A. Critères cliniques ........................................................................................................ 17
B. Phénotype tumoral ...................................................................................................... 19
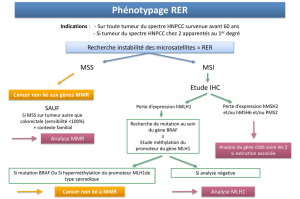
1. Recherche d’une instabilité microsatellitaire.............................................. 20
2. Analyse en immunohistochimie des protéines MMR ................................. 21
III. Génétique moléculaire ......................................................................................... 24
A. Place du diagnostic génétique ..................................................................................... 24
B. Les gènes MMR .......................................................................................................... 25
C. Types et répartitions des mutations............................................................................. 27
D. Méthodes d’identification ........................................................................................... 30
E. Diagnostics différentiels ............................................................................................. 30
F. Problèmes des variants de Signification Clinique et Biologique Inconnue (VSI) ...... 31
1. Définition .................................................................................................... 31
2. Outils pour l’interprétation des VSI ........................................................... 33
IV. Objectif du travail ................................................................................................ 35
Matériels et Méthodes................................................................................................. 36
I. Choix des mutations ............................................................................................. 36
II. Analyses in silico ................................................................................................... 37

III. Etude ex vivo de l’expression des variants ......................................................... 38
A. Obtention des constructions ........................................................................................ 40
1. Mutagenèse dirigée ..................................................................................... 40
2. Transformation bactérienne ........................................................................ 41
3. Extraction d’ADN ....................................................................................... 41
4. Séquençage ................................................................................................. 42
B. Transfection cellulaire ................................................................................................ 42
1. Lignées cellulaires et conditions de culture ................................................ 42
2. Transfection transitoire ............................................................................... 43
C. Analyse par Western blot ............................................................................................ 44
1. Préparation du lysat protéique .................................................................... 44
2. Migration électrophorétique et transfert des protéines sur membrane ....... 44
3. Immunomarquage et révélation .................................................................. 45
4. Interprétation............................................................................................... 46
Résultats ....................................................................................................................... 48
I. Caractéristiques cliniques des patients porteurs de VSI et phénotypes
tumoraux ............................................................................................................... 51
II. Données concernant l’impact des variants sur l’épissage ................................. 52
III. Analyses in silico de l’effet des variants sur la protéine ................................... 52
A. Variants MLH1 ............................................................................................................ 53
B. Variants MSH2 ............................................................................................................. 53
IV. Analyses ex vivo de l’expression des protéines MMR mutées .......................... 54
A. Obtention des constructions ......................................................................................... 54
B. Etudes des contrôles ..................................................................................................... 55
C. Etudes des VSI ............................................................................................................. 57
1. Variants MLH1 ........................................................................................... 57
2. Variants MSH2 ........................................................................................... 58
Discussion ..................................................................................................................... 61
Conclusion ................................................................................................................... 74
Références bibliographiques ...................................................................................... 75
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
1
/
94
100%