13 n° f i c h e

Edimark Santé, c’est aussi :
EDIMARK SAS
Les Lettres...
La Lettre du Cancérologue
La Lettre du Gynécologue
La Lettre de l’Hépato-gastroentérologue
La Lettre de l’Infectiologue
La Lettre du Neurologue
La Lettre d’Oto-rhino-laryngologie
et de chirurgie cervico-faciale
La Lettre du Pharmacologue
La Lettre du Pneumologue
La Lettre du Psychiatre
La Lettre du Rhumatologue
La Lettre du Sénologue
Les Correspondances...
Correspondances en Onco-Hématologie
Correspondances en Onco-Théranostic
Correspondances en Onco-Urologie
Correspondances en Métabolismes
Hormones Diabètes et Nutrition
Les Courriers...
Le Courrier des Addictions
Le Courrier de l’Algologie
Le Courrier de l’Éthique médicale
Le Courrier de la Transplantation
Les Images...
Images en Dermatologie
Images en Ophtalmologie
DaTeBe Éditions
Éditeur de livres
Collection “Urgences”
Collection “Les objectifs FMC”
Collection “Institutions et sociétés savantes”
Collection “La FMC tout en un”
Collection “Le pratique”
Collection “Santé au féminin”
À découvrir sur notre site !
www.edimark.fr
FICHE À DÉTACHER
La Lettre du Cardiologue • n° 458 - octobre 2012 | I
fiche
pratique
Sous la responsabilité de son auteur
n° 13
P. Charron*
* Centre de référence pour les maladies cardiaques héréditaires, hôpital de la Pitié-
Salpêtrière, AP-HP, Paris.
L
a découverte d’une cardiomyopathie dilatée
(CMD) conduit à réaliser un bilan étiologique afin
de rechercher une cause curable ou requérant un
traitement spécifique, d'adapter la surveillance médicale
du patient lorsque des complications particulières sont à
craindre, et de déterminer si une surveillance cardiologique
des apparentés est souhaitable.
Démarche générale
Après la mise en évidence d’un ventricule gauche dilaté
hypokinétique (fraction d'éjection ventriculaire gauche
inférieure à 45 % et diamètres ventriculaires gauches
télédiastolique et télésystolique supérieurs à 32mm/m2
ou au 95epercentile), la démarche vise à affirmer la CMD
en écartant les causes communes de dysfonction systolique
(maladie coronaire, hypertension artérielle, valvulopathies,
etc.), puis à envisager la recherche de causes particulières
à la CMD. Du fait de la multiplicité de causes sous-jacentes
possibles
(tableauI)
[1]
et du rendement souvent médiocre
du bilan étiologique, ce bilan initial ne peut être exhaustif
et il privilégie la recherche des pathologies avec traitement
spécifique. En revanche, l’analyse attentive du patient et
de son histoire familiale doit systématiquement rechercher
des signes d’appel. Ceux-ci vont amener à approfondir le
bilan étiologique, qui sera donc orienté au cas par cas.
Anamnèse, symptômes et examen physique
Les éléments atypiques à rechercher sont notamment
l’âge très précoce au diagnostic (maladies métaboliques),
l’exposition à des substances cardiotoxiques (médi caments,
alcool, etc.), un contexte fébrile (myocardite), des diffi-
cultés d’apprentissage (maladie de Steinert, maladies
mitochondriales, maladie de Danon), la présence d’une
surdité, d’un diabète, de troubles de la vision (maladies
mitochondriales), un déficit musculaire squelettique ou des
myotonies (maladie de Steinert, dystrophinopathies, lamino-
pathie, desminopathie), une hyperpigmentation cutanée
(hémochromatose), une cataracte précoce (desminopathie).
Une histoire familiale est présente dans un tiers des cas de
CMD et doit systématiquement être recherchée.
Électrocardiogramme
Les signes d’appel sur l’ECG sont le bloc auriculoventricu-
laire (myocardite, sarcoïdose, laminopathie, desminopathie),
Quel bilan étiologique
devantunecardiomyopathie dilatée ?
Tableau I. Principales causes de CMD.
Causes infectieuses, inflammatoires, immunes
◆ Virus (HIV, etc.), bactéries, mycobactéries, champignons, parasites
(chagas)
◆ Sarcoïdose, maladie de Kawasaki, syndrome de Churg-Strauss
Causes toxiques
◆ Alcool, anthracyclines, cobalt, agents antirétroviraux,
phénothiazines, plomb, mercure, cocaïne
Causes métaboliques
◆ Carences nutritionnelles (thiamine, sélénium, carnitine)
◆ Désordre électrolytique (hypocalcémie, hypophosphatémie)
◆ Causes endocriniennes (hypothyroïdie, acromégalie, thyrotoxicose,
maladie de Cushing, phéochromocytome, diabète)
Péripartum
Cardiopathie rythmique
Causes familiales/génétiques
◆ Avec myopathie : dystrophinopathie (Duchenne/Becker ),
dystrophie myotonique (Steinert), desminopathie (gène desmine),
laminopathie (gène des lamines A/C)
◆ Syndromique : cytopathie mitochondriale, syndrome de Barth
◆ Associant CMD et troubles conductifs : laminopathie
(gène des lamines A/C), desminopathie (gène desmine)
◆ CMD isolée : gènes du sarcomère, bande Z, cytosquelette,
desmosome

FICHE À DÉTACHER
II | La Lettre du Cardiologue • n° 458 - octobre 2012
fiche pratique n° 13
Tableau III. Bilan familial et génétique à préconiser.
Bilan cardiologique (ECG/échographie) chez les apparentés
◆ Préconisé chez tous les apparentés au 1er degré (fratrie, enfants,
parents), quel que soit le contexte familial (au moins à partir del’âge de
10 ans pour les enfants)
◆ S’il s’agit d’une forme familiale : préconiser la poursuite d’une surveil-
lance cardiologique régulière (échocardiographie), même en cas de
premier bilan normal, chez les apparentés au 1erdegré (tous les 1 à 2ans
entre l’âge de 10ans et celui de 20ans, puis tous les2 à 5ans entre
l’âge de 20ans et celui de 50à60ans) du fait de l’expression cardiaque
tardive. À adapter si les données génétiques sont disponibles (pas de
surveillance cardiaque si la mutation est absente chez l’apparenté)
Analyse génétique chez le patient avec CMD
◆ Proposée systématiquement en cas de forme familiale : gènes du
sarcomère (exemple : myosineβ) si le phénotype est commun (CMD),
ou autres gènes ciblés, si le phénotype est particulier
◆ Proposée en cas de CMD sporadique uniquement en cas de
phénotype particulier évocateur : troubles conductifs, myopathie ou CPK
élevée (gènes ciblés selon le phénotype et le bilan étiologique complet)
la pseudo-séquelle d’infarctus postéro-inférieur (sarcoïdose,
dystrophinopathie) et le faible voltage des QRS (mutation
phospholamban).
Échocardiographie
Le bilan étiologique doit être poussé en cas d’akinésie
ou de dyskinésie segmentaire (myocardite, sarcoïdose,
dystrophinopathie), d’hypertrophie des parois (les causes
d’hypertrophie, d'infiltration, de restriction, ou bien la
myocardite aiguë) ou de non-compaction du myocarde
(formes génétiques de CMD, notamment sarcomériques).
Biologie
Certains examens simples et pertinents
(tableauII)
doivent
systématiquement être demandés (une élévation chronique
de créatine phosphokinase (CPK) doit, par exemple, faire
rechercher une myopathie, une carence martiale évoque
une hémochromatose, une hyperéosinophile évoque un
syndrome de Churg-Strauss, une neutropénie évoque un
syndrome de Barth), alors que d’autres examens plus spécia-
lisés sont réalisés en cas de signes d’appel.
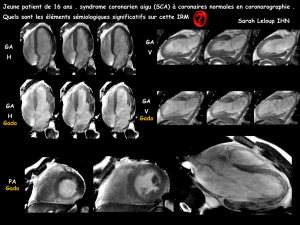
Imagerie par résonance magnétique
Les signes d’appel en IRM
(figure)
sont une anomalie de
la cinétique segmentaire (myocardite, sarcoïdose, dystro-
phinopathie), un aspect d’œdeme (T2) et de rehaussement
précoce (T1) [myocardite], un rehaussement tardif “en
mottes” à mi-paroi (postmyocardite, dystrophinopathie),
une anomalie de séquenceT2* (hémochromatose), un
remplacement adipeux intrapariétal (T1wFS) [“DVDA” du
ventricule gauche]
(2)
.
Biopsie
La biopsie endomyocardique est le “gold standard” pour
le diagnostic de myocardite et elle est utile au diagnostic
étiologique de nombreuses pathologies à l’origine d'une
CMD
(3)
. Elle est indiquée en présence de signes d’appel.
La biopsie musculaire squelettique peut constituer une
alternative en cas de stigmates musculaires, tels qu’une
élévation inexpliquée de CPK.
Bilan cardiologique familial
et test génétique
Le bilan cardiologique chez les apparentés est recommandé
devant toute CMD, et le test génétique est largement
préconisé, sauf en cas de cause acquise démontrée
(tableauIII)
[4]
. ■
1. Luk A, Ahn E, Soor GS, Butany J. Dilated cardiomyo-
pathy: a review. J Clin Pathol 2009;62:219-25.
2. Karamitsos TD, Francis JM, Neubauer S. The current
and emerging role of cardiovascular magnetic reso-
nance in the diagnosis of nonischemic cardiomyopa-
thies. Prog Cardiovasc Dis 2011;54:253-65.
3. Cooper LT, Baughman KL, Feldman AM et al. The
role of endomyocardial biopsy in the management of
cardiovascular disease: a scientific statement from the
American Heart Association, the American College of
Cardiology, and the European Society of Cardiology.
Circulation 2007;116:2216-33.
4. Charron P, Arad M, Arbustini E et al. European Society
of Cardiology Working Group on Myocardial and Peri-
cardial Diseases. Genetic counselling and testing in
cardiomyopathies: a position statement of the European
Society of Cardiology Working Group on Myocardial
and Pericardial Diseases. Eur Heart J 2010;31:2715-26.
Références bibliographiques
Tableau II. Bilan biologique à visée étiologique dans la CMD.
Bilan en première intention Bilan en seconde intention
◆ NFS plaquettes
◆ CPK (créatine phosphokinase)
◆ Calcémie, phosphatémie
◆ Fer sérique, ferritine
◆ TSH
(Thyroïd Stimulating
Hormone)
◆ Sérologie HIV
◆ CRP
(C Reactive Protein)
◆ Thiamine, carnitine, sélénium
◆ Sérologies virales, Lyme,
Chagas
◆ Catécholamines plasma/urines
◆ Enzyme de conversion
del’angiotensine
◆ Bilan immun (anticorps divers,
autoanticorps)
Figure. Exemples d’images par IRM (d’après
[2]
).
A : ventricule gauche dilaté à parois fines avec rehaussement tardif
à mi-paroi (flèches) typique d’une cardiomyopathie non ischémique.
B : rehaussement tardif de localisation multiple “en mottes”, typique
d’une myocardite.
A
B
1
/
2
100%