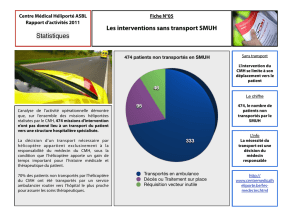
Mise au point clinique Comment la cardiomyopathie

12:48:15:04:13
Page 201
Page 201
Mise au point clinique
Comment la cardiomyopathie hypertrophique est devenue une
maladie actuelle, accessible au traitement
Barry J. Maron, MD ; Eugene Braunwald, MD
Observation clinique n° 1 : Le
patient est aujourd’hui âgé de
56 ans et présente une cardiomyopathie
hypertrophique (CMH) non obstruc-
tive. Lorsqu’il avait 35 ans, son frère
(alors âgé de 39 ans) est décédé
brutalement, l’autopsie ayant révélé
qu’il était atteint d’une CMH. Notre
patient a donc fait l’objet d’explora-
tions diagnostiques qui ont permis de
déceler une CMH non obstructive
avec, notamment, un septum inter-
ventriculaire d’une épaisseur de 31 mm.
En raison de ses antécédents familiaux
de mort subite (et de son importante
hypertrophie myocardique), ce sujet a
été équipé d’un défibrillateur auto-
matique implantable (DAI) dans un
but de prévention primaire de la mort
subite. Cinq ans plus tard, alors que le
patient dormait (à 1h00 du matin), son
DAI a aboli un épisode de fibrillation
ventriculaire (FV) et restauré le rythme
sinusal (Figure 1). Neuf ans se sont
écoulés sans nouvel incident, mais, à
50 ans, le patient a présenté, pendant
son sommeil, un épisode de FV sem-
blable au premier qui a de même été
aboli par un choc électrique. Aujour-
d’hui, à 56 ans, cet homme demeure
asymptomatique et actif. L’évolution
clinique de sa CMH a seulement
été ponctuée par ces deux épisodes
imprévisibles d’arrêt cardiaque,
efficacement jugulés par le DAI qui lui
avait été posé à titre préventif en raison
de son statut à haut risque.
Observation clinique n° 2 : La patiente
est une professeure des écoles âgée de
53 ans qui est atteinte de CMH obstruc-
tive. Elle a consulté à l’âge de 44 ans,
alors qu’elle était asymptomatique
mais présentait de lourds antécédents
familiaux de morts subites survenues
secondairement à une CMH chez deux
de ses frères (âgés de, respectivement,
20 et 34 ans). Un DAI lui a été implanté
à titre préventif et, deux ans plus tard
(la patiente étant âgée de 46 ans), celui-
ci a aboli une FV apparue pendant le
sommeil. Au cours des six années qui
ont suivi, quatre autres interventions
appropriées du DAI ont été nécessaires
pour juguler des épisodes de tachy-
cardie ventriculaire (TV) ou de FV.
L’épaisseur du septum interventri-
culaire est de 21 mm ; il n’existe pas
d’obstacle à l’éjection ventriculaire
gauche au repos, mais une obstruction
apparaît à l’effort (gradient de pres-
sion : 70 mmHg). A 51 ans, la dyspnée
d’effort et l’incapacité fonctionnelle
ont augmenté malgré une prise en
charge médicale maximale, avec un
handicap jugé de classe fonctionnelle
III de la New York Heart Association.
Une myectomie septale chirurgicale est
pratiquée, qui abolit le gradient de
pression et les symptômes d’insuf-
fisance cardiaque de la patiente. A 53
ans, cette femme est redevenue asymp-
tomatique et active après avoir fait
l’expérience de deux processus mor-
bides, à savoir le risque de mort subite
et l’insuffisance cardiaque secondaire
à l’obstacle à l’éjection ventriculaire
gauche, qui ont tous deux pu être
jugulés par la mise en œuvre de
stratégies thérapeutiques efficaces.
Introduction
La médecine cardiovasculaire progresse
rapidement, parfois même à la vitesse
de l’éclair, pour fournir de nouveaux
moyens thérapeutiques d’un apport
majeur. La maladie coronaire est au
cœur de la pratique cardiologique du
fait de son caractère épidémique actuel
et déjà au siècle dernier. Mais qu’en
est-il des affections cardiaques hérédi-
taires moins fréquentes, longtemps
négligées et qui, parfois, semblent se
perdre dans la masse des patients
atteints de cardiopathies ischémiques ?
La CMH est peut-être la plus
fréquente des maladies cardiaques
d’origine génétique.2,3 Nous allons voir
ici comment cette affection, autrefois
considérée comme aussi rare que
singulière, mais néanmoins intéres-
sante (et de sombre pronostic), est
désormais reconnue comme une entité
clinique relativement courante, actuelle
et accessible au traitement, ayant
donné lieu à des recommandations et
des préconisations fondées sur un
consensus (Figures 1 à 3).4–7 Cette
évolution est pleine d’enseignements
tant pour les médecins que pour leurs
patients atteints de CMH.
Histoire de la CMH
La CMH est aujourd’hui reconnue
comme la première cause de mort
subite chez le sujet jeune (y compris
l’athlète entraîné) ; elle est présente
chez au moins un individu sur 500 au
sein de la population générale et
affecterait quelque 750 000 Améri-
cains.8 Toutefois, avant de parvenir à
son actuel degré de reconnaissance, la
CMH a parfois connu un parcours
erratique et imprévisible au cours de
ses cinquante premières années.9,10 Ce
cheminement a été ponctué par une
myriade d’obstacles, liés, entre autre, à
Centre de prise en charge des cardiomyopathies hypertrophiques, Fondation de l’Institut cardiologique de Minneapolis, Minneapolis, Minnesota,
Etats-Unis (B.J.M.) ; et groupe d’étude TIMI, Brigham and Women’s Hospital, Boston, Massachusetts, Etats-Unis (E.B.).
Correspondance : Barry J. Maron, MD, Hypertrophic Cardiomyopathy Center, Minneapolis Heart Institute Foundation, 920 East 28th St, Suite 620,
Minneapolis, MN 55407, Etats-Unis. E-mail : hcm.maron@mhif.org
(Traduit de l’anglais : Evolution of Hypertrophic Cardiomyopathy to a Contemporary Treatable Disease. Circulation. 2012;126:1640–1644.)
© 2012 American Heart Association, Inc.
Circulation est disponible sur le site http://circ.ahajournals.org
201

12:48:15:04:13
Page 202
Page 202
de fausses conceptions et à de mauvais
débats auxquels est venue s’ajouter une
littérature souvent confuse et con-
tradictoire. Certains soutenaient
même au début (conceptions à présent
obsolètes) que la CMH ne constituait
pas une entité clinique spécifique ou
qu’il n’existait pas de réel obstacle
mécanique à l’éjection ventriculaire
gauche.9,10 Dans la littérature, une
litanie déconcertante de termes (75 au
total) était utilisée pour décrire la
même affection, dont ceux de
rétrécissement sous-aortique hyper-
trophique idiopathique et de cardio-
myopathie obstructive hypertrophique,
désormais remplacés par un seul, celui
de CMH.11
Au cours des premières années
(1960–1965), l’entité clinique jus-
qu’alors non reconnue que constituait
la CMH a fait l’objet de descriptions
cliniques aussi habiles qu’innovantes,
qui, pour une large part, ont d’abord
émané du centre clinique des National
Institutes of Health (NIH) des Etats-
Unis. Au départ, les possibilités de
traitement étaient quasi inexistantes.2
D’ailleurs, dans l’un des tout premiers
articles publiés sur cette affection à
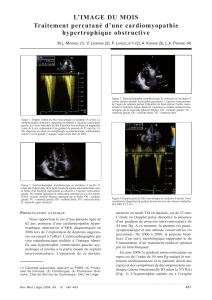
Figure 1. Prévention primaire de la mort subite par un DAI ayant aboli un épisode de TV-FV
à 41 ans (choc approprié n° 1). A, après 4 battements sinusaux, apparition brutale d’une TV
à 200 battements/min ; B, le défibrillateur détecte la TV et entame sa mise en charge d’une
durée de 8 secondes. C, la TV se dégrade en FV. D, le défibrillateur délivre un choc
approprié de 20 J (barre) et rétablit le rythme sinusal en un battement. MS : mort subite ;
DAI : défibrillateur automatique implantable ; TV : tachycardie ventriculaire ; FV : fibrillation
ventriculaire. Adapté et reproduit de Maron et al,1 avec l’autorisation de l’éditeur. Copyright ©
2007, American Medical Association.
l’instigation des NIH, décrivant
l’obstacle dynamique à l’éjection
ventriculaire gauche, toute tentative de
traitement était présentée comme
illusoire :12 «nous sommes conscients
qu’il n’existe à ce jour aucune
méthode de prise en charge capable
d’influer spécifiquement et favorable-
ment sur l’évolution d’un patient
atteint d’hypertrophie ventriculaire
idiopathique. »
Le constat était pleinement exact
à l’époque (c’est-à-dire en 1959), pour-
tant, peu de temps après, sont apparus
les bêtabloquants13,14 et la myectomie
chirurgicale.15 Les cardiologues ont
toutefois mis du temps à accepter ce
traitement invasif de la CMH. C’est
ainsi qu’au Royaume-Uni, après une
première expérience de courte durée, la
myectomie a été délaissée pendant
trente ans.
Un mythe désolant a pris corps, qui,
étonnamment, persiste dans certains
esprits, à savoir que la CMH était une
maladie implacable à laquelle il ne
pouvait être opposé aucune thérapeu-
tique curative ou efficace. La CMH a,
dès lors, acquis le statut d’affection
dont l’évolution était imprévisible et
aboutissait au décès dans la plupart des
cas. Le fait que cette conception
erronée continue à se perpétuer de nos
jours s’explique peut-être par les décès,
extrêmement rares mais néanmoins
très médiatisés, dont ont été victimes,
en pleine action, de jeunes athlètes
dont la CMH était jusqu’alors
méconnue.16
Les affections telles que la CMH
prêtent aux idées fausses. Les juge-
ments anciens imprègnent les con-
sciences et s’y enracinent, d’où la
difficulté de les faire disparaître même
en apportant des preuves contraires.
Les pathologies relativement rares en
pratique clinique, telle la CMH, con-
stituent des défis pour les cardiologues,
qui ont à cœur de servir au mieux leurs
patients, car elles leur imposent
d’acquérir suffisamment d’expérience
et de pratique pour être en mesure
d’apprécier pleinement les nuances de
leur expression clinique polymorphe et
de leur prise en charge.
La CMH de nos jours
Le profil clinique de la CMH et
l’arsenal des moyens de traitement ont
considérablement évolué. En premier
lieu, on s’accorde désormais pour con-
sidérer que la plupart des individus
atteints d’une forme génétique de la
maladie ne font probablement l’objet
d’aucun diagnostic clinique, nombre
d’entre eux parvenant jusqu’à l’âge
de soixante-dix, quatre-vingts, voire
quatre-vingt-dix ans, sans éprouver de
handicap ni avoir eu à subir des inter-
ventions thérapeutiques majeures.4–7,17
En effet, du fait, principalement, de la
tendance accrue à suspecter la maladie
qui découle en partie de la dissémi-
nation des techniques d’imagerie de
pointe, chez les patients dont la CMH
est asymptomatique, le diagnostic
fortuit de l’affection est plus souvent
fait à un âge relativement avancé.
En outre, le risque de complications
liées à la CMH (dont celui de mort
subite) est paradoxalement faible chez
les patients âgés de plus de 60 ans.18
Pour autant, aujourd’hui encore, la
plupart des patients chez lesquels une
CMH vient d’être découverte semblent
surpris (et rassurés) d’apprendre cet
202 Circulation Mai 2013

12:48:15:04:13
Page 203
Page 203
important principe de l’effet du vieil-
lissement sur la maladie, cela en raison
du message généralement défavorable
qui leur a été délivré concernant leur
état.
Par ailleurs, nombre de patients qui
consultent dans un hôpital ou un centre
de soins présentent (ou encourent)
des complications inhérentes à leur
CMH1,4–7,19–21 (Figure 3). L’évolution
clinique de la maladie peut se faire
dans plus d’une direction défavorable,
conduisant alors à employer des
moyens thérapeutiques habituellement
réservés aux patients atteints d’autres
pathologies cardiaques (Figure 3). Ces
complications imposent des stratégies
de prise en charge spécifiques ; en
Figure 2. Retentissement clinique de l’obstacle à l’éjection VG dans la CMH. Graphique
du haut, les patients qui présentent un tel processus obstructif au repos (gradient de
pression ≥30 mmHg) encourent un plus grand risque d’insuffisance cardiaque progressive
sévère ou de décès par insuffisance cardiaque ou accident vasculaire cérébral que ceux
dont la maladie n’entraîne pas d’obstacle à l’éjection. CMH : cardiomyopathie
hypertrophique ; VG : ventriculaire gauche ; NYHA : New York Heart Association ; RR : risque
relatif. Reproduit avec l’autorisation de Maron et al.19 Graphique du bas, la myectomie
septale chirurgicale, qui a pour effet d’abolir le gradient de pression VG, assure aux patients
une espérance de vie à long terme comparable à celle dont est créditée la population
générale des Etats-Unis et qui excède celle des individus atteints de CMH obstructive
symptomatique n’ayant pas été opérés. Reproduit d’Ommen et al20 avec l’autorisation de
l’éditeur. Copyright © 2005, New England Journal of Medicine.
principe, il ne saurait être proposé de
traitement unique à tous les patients,
tant la maladie est hétérogène.
Prévention de la mort subite
La mort subite est la complication la
plus spectaculaire et la plus terrible de
la CMH, connue depuis la première
description anatomopathologique
publiée en 1958.3 Pour autant, la
prévention de la mort subite est
aujourd’hui devenue un objectif réa-
liste chez le jeune patient atteint de
CMH.1,21 Si la mort subite de patients
jeunes a longtemps marqué l’histoire
de cette affection du fait de l’absence
de traitement capable de prévenir ce
risque, une stratégie de prévention
systématique s’est progressivement
imposée au cours des dix dernières
années, visant à protéger ces patients
par la pose d’un DAI.1,21
Sa capacité à déceler et abolir
automatiquement les TV et FV fait du
DAI la seule approche thérapeutique
aujourd’hui disponible pour prolonger
de façon effective la vie des individus
atteints de CMH, permettant, par là
même, d’infléchir le cours naturel de la
maladie chez de nombreux patients
(Figure 1). Lorsque les DAI sont
employés dans la CMH, y compris
chez les patients à haut risque, leur
taux annuel d’intervention à des fins de
prévention primaire est de 4 %, tant
chez l’adulte que chez l’enfant.1 Bien
que le rôle salvateur du DAI soit
pleinement établi dans la CMH, le
problème demeure d’identifier de façon
plus précise les patients susceptibles de
tirer profit de ce traitement, notam-
ment au sein du sous-groupe, restreint
mais néanmoins important, formé par
les individus chez lesquels les habituels
marqueurs de risque n’ont pas de
valeur prédictive, mais qui peuvent
malgré tout être un jour victimes d’une
mort subite.21 C’est pourquoi le
dépistage d’un plus grand nombre de
patients à risque par l’emploi de
nouveaux marqueurs demeure un
objectif important, dans le cadre
duquel l’IRM cardiovasculaire de
contraste avec recherche d’un
rehaussement tardif du gadolinium
(supposé témoigner de la présence
d’une cicatrice myocardique) est
appelée à jouer un rôle significatif.22
Abolition de l’insuffisance cardiaque
Dans la CMH, l’insuffisance cardiaque
sévère et l’intolérance à l’effort
imputables à l’obstacle à l’éjection
ventriculaire gauche sont potentielle-
ment réversibles20 (Figure 2). La
myectomie septale chirurgicale, dont
le pionnier a été Andrew Morrow des
NIH23 (ensuite améliorée par d’autres,
dont Bill Williams de Toronto, John
Kirklin et maintenant Joseph Dearani,
tous deux membres de la Mayo Clinic),
est aujourd’hui devenue une inter-
vention sûre, qui restaure ad integrum
la qualité de vie des patients en
Maron et Braunwald La CMH est une maladie actuelle, accessible au traitement 203

12:48:15:04:13
Page 204
Page 204
normalisant les pressions ventriculaires
gauches.
A l’origine, même pratiquée par les
mains les plus expérimentées, la myec-
tomie était grevée d’une mortalité
opératoire comprise entre 5 et 8 %, ce
qui contribuait incontestablement à
tempérer l’attrait pour cette inter-
vention. Aussi le nombre de myec-
tomies réalisées demeurait relativement
faible.
Il a fallu attendre ces dix à quinze
dernières années pour que soient
pleinement reconnues la fréquence et
l’importance du facteur obstructif
dans le spectre clinique de la CMH19,24
(Figure 2). La myectomie septale
chirurgicale est alors devenue une
option séduisante et accessible, comme
en témoigne l’augmentation spectacu-
laire du nombre d’interventions
pratiquées annuellement dans les
grands centres.25,26 Ce regain d’intérêt
pour la myectomie est dû à l’abaisse-
ment considérable de la mortalité
opératoire à 1 % ou moins, rendu
possible par les techniques modernes
de préservation myocardique, le gain
d’expérience des chirurgiens et les
innovations apportées à cette tech-
nique opératoire.25,26 C’est ainsi qu’a
été développée l’approche désignée
sous le nom de myectomie étendue,
pour prendre en compte le rôle
important joué par les structures sous-
mitrales dans l’obstacle à l’éjection
Figure 3. Evénements susceptibles de compliquer la CMH. Chacune de ces complications
de la maladie est accessible à au moins une option thérapeutique potentiellement efficace.
AVC : accident vasculaire cérébral ; FA : fibrillation atriale ; DAI : défibrillateur automatique
implantable ; CMH : cardiomyopathie hypertrophique ; ARF : ablation par radiofréquence.
ventriculaire gauche.4–7,27 Surtout, en
plus de lever cet obstacle et d’améliorer
par là même la qualité de vie des
patients, la myectomie chirurgicale
augmente également l’espérance de vie
de ces derniers, la ramenant au niveau
de celle de la population générale et au-
delà de celle des patients atteints d’une
CMH obstructive symptomatique qui
n’ont pas été opérés20 (Figure 2).
Un mode d’intervention par voie
percutanée, l’ablation septale par
alcoolisation, constitue un important
substitut à la myectomie,5–7,28 toutefois
réservé aux patients trop âgés et/ou
considérés comme de moins bons
candidats à l’approche chirurgicale en
raison de comorbidités ou de leur forte
aversion pour la chirurgie.4–7
Si l’alcoolisation septale est recon-
nue quant à son efficacité à réduire le
gradient de pression et les symptômes,
elle provoque toutefois un infarctus
myocardique transmural lié à l’action
de l’alcool et qui est potentiellement
arythmogène, ce qui la distingue
notablement de la myectomie. Pour
autant, l’enthousiasme avec lequel les
cardiologues interventionnels ont
adopté l’alcoolisation septale a
renforcé la sensibilisation à l’égard
de la CMH et a, paradoxalement,
accentué l’intérêt porté à l’abolition
de l’obstacle à l’éjection ventricu-
laire gauche par réalisation d’une
myectomie.25,26,28,29
Les rares patients à présenter une
insuffisance cardiaque très évoluée,
réfractaire au traitement médica-
menteux et à caractère irréversible,
généralement liée à l’existence d’une
dysfonction systolique et d’un remode-
lage ventriculaire gauche délétère,
eux-mêmes secondaires à une ischémie
microvasculaire et à une cicatrice
diffuse (stade terminal), relèvent
d’une greffe cardiaque, seule option
thérapeutique efficace dans leur cas.30
Chez ces patients atteints de CMH, la
survie après transplantation est du
même ordre que celle des individus
opérés pour d’autres affections car-
diaques : elle est de 85 % à 1 an, de
75 % à 5 ans et de 61 % à 10 ans,
contre, respectivement, 82, 70 et 49 %
pour les transplantés non opérés pour
une CMH.31
Fibrillation atriale et accident
vasculaire cérébral
La fibrillation atriale (FA) est le plus
fréquent des troubles du rythme
soutenus associés à la CMH, survenant
chez environ 20 % des patients (ce qui
est quatre fois supérieur au taux
observé dans la population générale).32
Chez certains patients, la FA peut être
cause d’une insuffisance cardiaque
progressive ou d’un accident vasculaire
cérébral d’origine embolique (mais n’a
toutefois pas d’incidence sur le risque
de mort subite inattendue). Les stra-
tégies de prise en charge de la FA sont
les mêmes chez les patients atteints de
CMH que chez les autres individus
et font preuve d’une efficacité com-
parable. Les thérapeutiques pharma-
cologiques conventionnelles sont
administrées selon des protocoles
agressifs avec pour objectifs de
diminuer la fréquence des épisodes
paroxystiques, de maintenir le rythme
sinusal, de normaliser la fréquence
ventriculaire et de prévenir les acci-
dents vasculaires cérébraux d’origine
embolique par l’instauration d’une
anticoagulation à visée préventive.
Certains patients dont la FA se montre
réfractaire au traitement médica-
menteux peuvent se voir proposer une
ablation par courant de radiofréquence
destinée à abolir le foyer de FA,
204 Circulation Mai 2013

12:48:15:04:13
Page 205
Page 205
intervention qui, lorsqu’elle est pra-
tiquée précocement, permet de restau-
rer le rythme sinusal et d’améliorer la
symptomatologie à court terme chez
plus de 50 % des patients.33
Conclusions et perspectives
La CMH est sortie d’une période de
profonde incompréhension, de rejet
et de pessimisme pour devenir une
affection cardiovasculaire actuelle dont
chacune des morbidités et des compli-
cations peut être prise en charge par
des stratégies largement répandues et
efficaces. Ces moyens thérapeutiques
majeurs dont les patients atteints de
CMH sont à même de bénéficier ont la
capacité de modifier le cours naturel de
la maladie, notamment en prévenant
le risque de mort subite (par la pose
d’un DAI) et en faisant régresser
l’insuffisance cardiaque, avec pour
corollaires la restauration de la qualité
de vie et l’allongement de l’espérance
de vie (grâce à la myectomie
chirurgicale).
Il convient de souligner que les
stratégies matures de prise en charge de
la CMH dont il a été traité ici cons-
tituent les applications directes de
principes et de travaux de recherche
purement cliniques et ne doivent rien
aux importants progrès accomplis dans
le domaine la biologie moléculaire. En
effet, si ces derniers ont effectivement
permis d’identifier les mutations
génétiques codant pour les protéines
sarcomériques responsables de CMH,
identification dont l’impact clinique se
limite pour l’essentiel au dépistage
familial,34 ils n’ont, jusqu’à présent,
joué aucun rôle dans le traitement et
l’évaluation pronostique des CMH
ayant une expression clinique.
Beaucoup de travail reste à faire sur
cette affection génétique complexe,
notamment sur le plan de la stratifica-
tion du risque. Pour autant, l’objectif
final, à savoir permettre aux patients
atteints d’une CMH de bénéficier
d’une espérance de vie normale et
d’une qualité de vie satisfaisante, est
aujourd’hui devenu une aspiration
réaliste grâce aux importants efforts
déployés par de nombreux médecins
et investigateurs cliniques depuis la
reconnaissance initiale de cette maladie
il y a plus d’un demi-siècle.
Déclarations
Le Dr Maron est consultant auprès
de GeneDx et bénéficie d’une bourse de
recherche de Medtronic. Le Dr Braunwald
n’a aucun conflit d’intérêts à signaler.
Références
1. Maron BJ, Spirito P, Shen W-K, Haas TS,
Formisano F, Link MS, Epstein AE,
Almquist AK, Daubert JP, Lawrenz T,
Boriani G, Estes NAM 3rd, Favale S,
Piccininno M, Winters SL, Santini M,
Betocchi S, Arribas F, Sherrid MV, Buja G,
Semsarian C, Bruzzi P. Implantable cardio-
verterdefibrillators and prevention of
sudden cardiac death in hypertrophic
cardiomyopathy. JAMA. 2007;298:405–412.
2. Braunwald E, Lambrew CT, Rockoff SD,
Ross J Jr, Morrow AG. Idiopathic hyper-
trophic subaortic stenosis. I. A description
of the disease based upon an analysis of 64
patients. Circulation. 1964;30:3–119.
3. Teare D. Asymmetrical hypertrophy of the
heart in young adults. Br Heart J. 1958;20:
1–8.
4. Maron BJ, McKenna WJ, Danielson GK,
Kappenberger LJ, Kuhn HJ, Seidman CE,
Shah PM, Spencer WH, Spirito P, ten Cate
FJ, Wigle ED. American College of
Cardiology/European Society of Cardio-
logy Clinical Expert Consensus Document
on Hypertrophic Cardiomyopathy. J Am
Coll Cardiol. 2003;42:1687–1713.
5. Gersh BJ, Maron BJ, Bonow RO,
Dearani JA, Fifer MA, Link MS, Naidu SS,
Nishimura RA, Ommen SR, Rawkowski H,
Seidman CE, Towbin JA, Udelson JE,
Yancy CW; American College of Cardio-
logy Foundation/American Heart Asso-
ciation Task Force on Practice Guidelines;
American Association for Thoracic Surgery;
American Society of Echocardiography;
American Society of Nuclear Cardiology;
Heart Failure Society of America; Heart
Rhythm Society; Society for Cardiovascular
Angiography and Interventions; Society
of Thoracic Surgeons. 2011 ACCF/AHA
guidelines for the diagnosis and treatment
of hypertrophic cardiomyopathy: a report
of the American College of Cardiology
Foundation/American Heart Association
Task Force on Practice Guidelines.
Circulation. 2011;124:e783–e831.
6. Gersh BJ, Maron BJ, Bonow RO,
Dearani JA, Fifer MA, Link MS, Naidu SS,
Nishimura RA, Ommen SR, Rawkowski H,
Seidman CE, Towbin JA, Udelson JE,
Yancy CW; American College of
Cardiology Foundation/American Heart
Association Task Force on Practice Guide-
lines. 2011 ACCF/AHA Guidelines for the
Diagnosis and Treatment of Hypertrophic
Cardiomyopathy: a report of the American
College of Cardiology Foundation/Ameri-
can Heart Association Task Force on Prac-
tice Guidelines. Developed in collaboration
with the American Association for Thoracic
Surgery, American Society of Echocar-
diography, American Society of Nuclear
Cardiology, Heart Failure Society of
America, Heart Rhythm Society, Society for
Cardiovascular Angiography and Inter-
ventions, and Society of Thoracic Surgeons.
J Am Coll Cardiol. 2011;5:e212–e260.
7. American College of Cardiology Founda-
tion/American Heart Association Task
Force on Practice; American Association for
Thoracic Surgery; American Society of
Echocardiography; American Society of
Nuclear Cardiology; Heart Failure Society
of America; Heart Rhythm Society; Society
for Cardiovascular Angiography and Inter-
ventions; Society of Thoracic Surgeons,
Gersh BJ, Maron BJ, Bonow RO,
Dearani JA, Fifer MA, Link MS, Naidu SS,
Nishimura RA, Ommen SR, Rakowski H,
Seidman CE, Towbin JA, Udelson JE,
Yancy CW. 2011 ACCF/AHA guidelines for
the diagnosis and treatment of hypertrophic
cardiomyopathy: a report of the American
College of Cardiology Foundation/
American Heart Association Task Force on
Practice Guidelines. J Thorac Cardiovasc
Surg. 2011;142:e153–e203.
8. Maron BJ, Gardin JM, Flack JM,
Gidding SS, Bild D. Assessment of the
prevalence of hypertrophic cardiomyopathy
in a general population of young adults:
Echocardiographic analysis of 4111 subjects
in the CARDIA Study. Circulation. 1995;92:
785–789.
9. Maron BJ, Maron MS, Wigle ED,
Braunwald E. The 50-year history,
controversy, and clinical implications of
left ventricular outflow tract obstruction
in hypertrophic cardiomyopathy: from
idiopathic hypertrophic subaortic stenosis
to hypertrophic cardiomyopathy. J Am Coll
Cardiol. 2009; 54:191–200.
10. Maron BJ, Braunwald E. Eugene
Braunwald MD and the early years of
hypertrophic cardiomyopathy: a conversa-
tion with Dr. Barry J. Maron. Am J Cardiol.
2012;109:1539–1547.
11. Maron BJ, Seidman CE, Ackerman MJ,
Towbin JA, Maron MS, Ommen SR,
Nishimura RA, Gersh BJ. What’s in a
name? Dilemmas in nomenclature charac-
terizing hypertrophic cardiomyopathy and
left ventricular hypertrophy. Circ Cardiovasc
Genet. 2009;2:81–86.
12. Morrow AG, Braunwald E. Functional
aortic stenosis: a malformation characte-
rized by resistance to left ventricular outflow
without anatomic obstruction. Circulation.
1959;20:181–189.
13. Harrison DC, Braunwald E, Glick G,
Mason DT, Chidsey CA, Ross J Jr. Effects
of betaadrenergic blockade on the circula-
tion with particular reference to observa-
tions in patients with hypertrophic subaortic
stenosis. Circulation. 1964;29:84–98.
14. Cohen LS, Braunwald E. Amelioration of
angina pectoris in idiopathic hypertrophic
Maron et Braunwald La CMH est une maladie actuelle, accessible au traitement 205
 6
6
1
/
6
100%