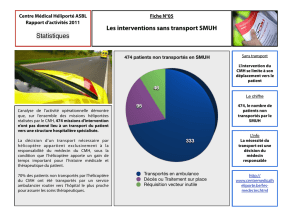
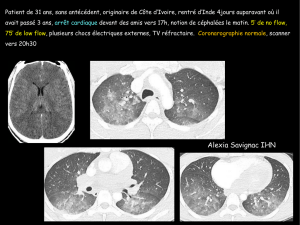
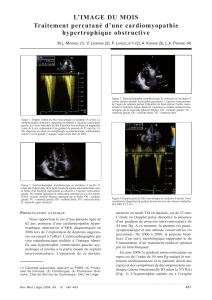
Actualités thérapeutiques dans la cardiomyopathie

1
Actualités thérapeutiques dans la cardiomyopathie hypertrophique
Marie L. Moonen (1), Luc A. Piérard (2).
AUTEURS :
(1) Aspirant F.R.S.-FNRS, candidat spécialiste. Service de Cardiologie, CHU Sart Tilman
B35, 4000 Liège. Tel + 3243667194, Fax +3243667195, [email protected]
(2) Professeur Ordinaire, Chef de Service. Service de Cardiologie, CHU Sart Tilman B35,
4000 Liège. Tel + 3243667194, Fax +3243667195, [email protected]
AUTEUR CORRESPONDANT :
Professeur Luc A. Piérard, Service de Cardiologie, CHU Sart Tilman B35, 4000 Liège.
Tel + 3243667194, Fax +3243667195, [email protected]
NOMBRE DE CARACTERES ESPACES COMPRIS : 14 639

2
RESUME
Longtemps considérées comme des pathologies rares, les cardiomyopathies
hypertrophiques (CMH) apparaissent actuellement, grâce à la généralisation des examens
échocardiographiques, nettement plus prévalentes. Spécifiquement, la démarche clinique se
doit d’être orientée en fonction de l’identification de groupes à risque. Le traitement médical
possède une place importante et est fréquemment sous maximal. Une prise en charge
interventionnelle est l’option thérapeutique réservée aux patients présentant une CMH
obstructive et qui conservent des symptômes sévères sous traitement médical maximal. La
décision d’implantation d’un défibrillateur automatique reste controversée en prévention
primaire.
SUMMARY. New trends in treatment of hypertrophic cardiomyopathy.
The management of patients with hypertrophic cardiomyopathy has largely evolved
over the past two decades. One important finding is that medical treatment appears under
used. Invasive procedure are reserved to patients with obstructive hypertrophic
cardiomyopathy who remain symptomatic despite optimal medical treatment. Indications for
implantable cardiac defibrillator are still debated. A global and multidisciplinary approach of
the patient and of his family is mandatory.

3
INTRODUCTION
Le diagnostic de cardiomyopathie hypertrophique (CMH) se pose habituellement par
l’identification d’une hypertrophie ventriculaire gauche (VG) en l’absence d’étiologie patente.
Cette définition, d’application clinique difficile car faisant référence à l’organisation
histologique du myocarde pathologique, a récemment été mise à jour par la société
européenne de cardiologie qui propose une classification des cardiomyopathies à partir des
anomalies morphologiques macroscopiques constatées, distinguant les CMH, les
cardiomyopathies dilatées, les cardiomyopathies arythmogènes du ventricule droit et les
cardiomyopathies restrictives.1 Chacune de ces catégories est subséquemment divisée en
deux, différenciant les formes génétiques/familiales des formes non génétiques/non familiales
(idiopathiques ou acquises) (tableau 1). L’application de cette nouvelle classification au
champ des CMH en a modifié quelque peu la définition : les CMH sont actuellement définies
par la présence d’une augmentation de l’épaisseur pariétale ou de la masse VG en l’absence
d’élévation des conditions de charge suffisante pour expliquer le degré d’hypertrophie
constatée. L’implication pratique de cette approche est de mieux coller à la pratique
quotidienne, de sensibiliser les praticiens à l’existence d’une potentielle origine génétique à
toute atteinte cardiaque et, finalement, d’offrir un cadre de travail dans lequel organiser
l’exploration complémentaire.
Une hypertrophie VG, en l’absence d’hypertension ou de valvulopathie, est constatée
chez approximativement 1:500 dans la population générale.2 Les formes familiales de CMH
représentent 55% des cas et une étiologie génétique doit être recherchée même dans les
formes sporadiques. Lorsque l’atteinte est familiale, il s’agit d’un mode de transmission
autosomique dominant. Toutefois, la présentation clinique de la maladie dépend, non
seulement de la mutation impliquée, mais également de facteurs environnementaux traduisant
une pénétrance variable responsable de l’expression phénotypique différente d’une même

4
mutation au sein des membres d’une famille. Classiquement, l’hypertrophie est alors
asymétrique. En cas d’hypertrophie VG concentrique, l’étiologie est, de préférence, une
atteinte métabolique, une mitochondriopathie ou une anomalie du stockage du glycogène. Le
mode de transmission, la distribution de l’hypertrophie et l’atteinte syndromique
éventuellement associée représentent autant d’éléments additionnels orientant le diagnostic
étiologique.
ASPECTS GENETIQUES ET IMPLICATIONS THERAPEUTIQUES
Au cours des 20 dernières années, de substantiels progrès ont été réalisés dans la
caractérisation des gènes impliqués dans la pathogenèse des CMH. Plusieurs centaines de
mutations sur plus de 27 gènes présumés sont actuellement décrites et impliquent, non
seulement des protéines structurelles des sarcomères ou des disques Z, mais également des
protéines des mitochondries et du métabolisme myocardique du calcium. Outre les
implications directes que la découverte de ces gènes offre à l’approche physiopathologique
des CMH, d’importantes implications cliniques, diagnostiques et thérapeutiques, en émanent
par l’intermédiaire d’un élargissement du dépistage génétique et de leur valeur pronostique
éventuelle associée.
Le dépistage génétique des CMH représente actuellement un outil à la disposition des
cliniciens. Un screening des mutations putatives les plus fréquemment impliquées est
réalisable chez le probant en quelques mois dans les centres spécialisés. Les deux mutations
les plus fréquemment rencontrées sont MYBPC3 (cardiac myosin-binding protein C) et
MYH7 (beta-myosin heavy chain) dont la prévalence est estimée entre 15 et 25% pour
chacune d’entre elles.3 Les implications psychologiques, éthiques et légales inhérentes à la
réalisation de tout dépistage doivent avoir fait l’objet, préalablement, d’une concertation avec

5
le probant et sa famille, de sorte qu’une prise en charge pluridisciplinaire est généralement
primordiale dans ces circonstances.
IDENTIFICATION DES GROUPES A RISQUE
La CMH représente une affection cardiologique unique dont les symptômes peuvent
se manifester de l’enfance à un âge de plus de 90 ans. Longtemps considérée comme une
affection grevée d’un pronostic péjoratif, les données actuelles sont en faveur d’une
réorientation du pronostic des patients porteurs de l’affection, indiquant une mortalité
annuelle de l’ordre d’1%/an, proche de la mortalité constatée dans la population générale.4
Ces observations recadrent la prise en charge de la maladie. L’objectif premier est de ne pas
nuire au patient en l’exposant à une prise en charge inutilement invasive et risquée face à une
histoire naturelle de bon pronostic, avec plus de 55% des patients présentant une forme a- ou
paucisymptomatique et dont l’espérance de vie est normale. Néanmoins, parmi le spectre
clinique de la CMH, il existe des groupes à plus haut risque d’événements cardiovasculaires
qu’il convient d’identifier. La mort subite représente l’un d’eux, non seulement le plus
dramatique car survenant principalement entre 20 et 30 ans, mais également le plus
difficilement prévisible. Les autres modes de présentation clinique aggravant le pronostic
comprennent le développement d’une insuffisance cardiaque et la survenue d’événements
cardio-emboliques à la faveur d’accès de fibrillation auriculaire (FA). La prise en charge se
doit d’identifier précisément ces patients afin qu’ils bénéficient d’un suivi rapproché et d’une
prise en charge orientée.
PRISE EN CHARGE DES PATIENTS À HAUT RISQUE DE MORT SUBITE
Alors que la mortalité annuelle constatée est de l’ordre d’1% par an dans la population
des patients porteurs d’une CMH, le risque de mort subite demeure important dans la tranche
 6
7
8
9
10
11
12
13
14
15
16
17
6
7
8
9
10
11
12
13
14
15
16
17
1
/
17
100%