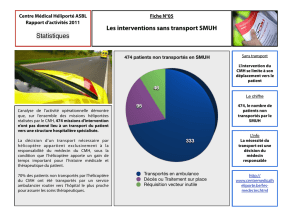
Fiche enseignant Les modèls animaux

La CardioMyopathie Hypertrophique familiale (CMH)
Les modèles animaux
Informations utilisables par les élèves :
La maladie humaine : Recherche Internet Wikipedia, Site Cardiomyopathie…
Comment fabriquer une souris transgénique ?
Résumé (en anglais) d’un article de la revue Science :
« A mouse model of familial hypertrophic cardiomyopathy (FHC) was generated by the introduction of an Arg403
Gln mutation into the cardiac myosin heavy chain (MHC) gene. Homozygous MHC403/403 mice died 7 days after
birth, and sedentary heterozygous MHC403/+ mice survived for 1 year. Cardiac histopathology and dysfunction in the
MHC403/+ mice resembled human FHC. Cardiac dysfunction preceded histopathologic changes, and myocyte
disarray, hypertrophy, and fibrosis increased with age. Young male MHC403/+ mice showed more evidence of disease
than did their female counterparts. Preliminary results suggested that exercise capacity may have been
compromised in the MHC403/+ mice. This mouse model may help to define the natural history of FHC. »
Si les élèves ont réalisé avant la séquence Relations Génotype – Phénotypes, ils devraient pouvoir
s’approprier ce texte. En cas de difficulté, il existe des systèmes de traduction automatique (par exemple
Google, menu Plus / traduction) qui donnent des résultats approximatifs. On peut, à titre d’exercice,
demander aux élèves de corriger les approximations de la traduction.
Observation du modèle murin
Cette premier modèle murin (pour la chaîne lourde béta de la myosine) a permis les observations suivantes :
Les souriceaux transgéniques homozygotes meurent environ une semaine après leur naissance
d'insuffisance cardiaque
Les souriceaux transgéniques hétérozygotes sont viables
A cinq semaines, leur fonction diastolique est altérée et leur débit cardiaque réduit.
A quinze semaines, des altérations de certaines cellules myocardiques apparaissent.
A six mois, ils présentent une hypertrophie cardiaque.
Le phénotype CMH est plus marqué chez les mâles que chez les femelles.
Validité du modèle biologique créé :
Similitudes avec la CMH humaine
Différences avec la CMH humaine
Fonction diastolique altérée
Influence du sexe de l'individu
Hypertrophie cardiaque
Dilatation auriculaire
Altération de la structure des cellules
Homozygotes létaux
-
Pas de mort subite
Source :
Geisterfer-Lowrance AA, Christe M, Conner DA, Ingwall JS, Schoen FJ, Seidman CE, Seidman JG.
A mouse model of familial hypertrophic cardiomyopathy.
Science. 1996 May 3;272(5262):731-4.
Intérêt du modèle murin
L'objectif des chercheurs est double : mieux comprendre la maladie et rechercher des traitements.
La transgenèse permet d'obtenir rapidement un grand nombre d'individus génétiquement identiques. Ils
peuvent être élevés dans un environnement rigoureusement contrôlé.

CMH Homme
CMH Souris transgénique
Génotype
Variable d'un malade à
l'autre
Identique entre individus
Environnement
Variable d'un malade à
l'autre
Maîtrisé par
l'expérimentateur
Durée d'une génération
Environ 20 ans
Quelques semaines
Nombre de sujets disponibles pour analyse
statistique
Limité
Illimité
Perspectives thérapeutiquesPour des raisons évidentes, la recherche de traitements pour les malades
atteints de CMH sera facilitée par l'existence de modèles biologiques.
Actuellement, les traitements utilisés ou envisagés sont les suivants :
Traitements médicamenteux : médicaments ralentissant ou régularisant le rythme cardiaque, ou
améliorant le remplissage cardiaque (béta-bloquants, antagonistes du calcium...)
Traitements chirurgicaux : suppression de la zone hypertrophiée par ablation
Infarctus contrôlé : Injection d'alcool à 95° dans l'artère coronaire irriguant la zone hypertrophiée, ce
qui provoque un infarctus localisé
Utilisation d'un stimulateur cardiaque (pace-maker) ou d'un défibrillateur
Transplantation cardiaque (dans les cas extrêmes)
Thérapie génique : perspective lointaine (problèmes éthiques, multitude des gènes impliqués...)
1
/
2
100%