P HARMACOMETRIE

L2 Pharmacie - Pharmacologie N° 17
06/04/14 - P. Boulouard
Groupe 35 - Anais et Lucas
1
PHARMACOMETRIE
II. Paramètres fonctionnels ..................................................................................................................... 2
III. Sélectivité ............................................................................................................................................... 11
IV. Notion de synergie additive ou potentialisatrice .................................................................. 12
V. Notion de marge thérapeutique ................................................................................................... 13
VI. Variabilité pharmacologique.......................................................................................................... 14

L2 Pharmacie - Pharmacologie N° 17
06/04/14 - P. Boulouard
Groupe 35 - Anais et Lucas
2
I. Interaction ligand-récepteur : notion d’affinité
II. Paramètres fonctionnels
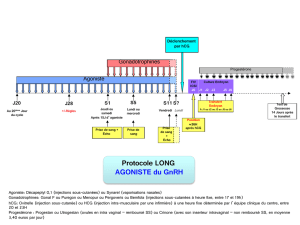
Agonistes, agonistes partiels, antagonistes
Cette représentation indique qu’il existe ;
- agoniste entier avec effet très important
- agoniste partiel où l’effet va être moins important que celui entrainé avec la fixation sur son
Rc de l’agoniste entier
- antagoniste neutre : qui n’a pas d’activité cellulaire
- agoniste inverse qui exerce effet inverse des agonistes entiers ou partiels avec une amplitude
qui peut être différente (entier ou partiel)
Ligand qui se fixe sur des Rc muscariniques :
Rc muscariniques sont les Rc sur lesquels se fixe
l’Acétylcholine (agit sur des Rc muscarinique M et des Rc
nicotiniques N), ils sont couplés aux protéines G et donc on va
mesurer expérimentalement l’activité GTPasique en
pourcentage
Le carbacol est un agoniste totale entier vis à vis des Rc
muscarinique en entrainant une activité maximale.
L’atropine qui est un agoniste neutre n’entraine pas de
modification de cette activité GTPasique
La pirenzépine agoniste inverse partiel entraine une
diminution de cette activité GTPasique
19
II-Paramètres fonctionnels
Agonistes, agonistes partiels, antagonistes
19
II-Paramètres fonctionnels
Agonistes, agonistes partiels, antagonistes

L2 Pharmacie - Pharmacologie N° 17
06/04/14 - P. Boulouard
Groupe 35 - Anais et Lucas
3
Réponse quantale
Principe du « tout ou rien » : une molécule
produit un effet ou pas. On définît le DE50
comme la dose nécessaire pour produire un effet
chez 50% des animaux
Mise au point molécule à visée hypnotique
(induire le sommeil) où l’on fait des lots de 10
souris avec différentes concentrations. On
injecte une dose à chaque lot et on mesure
l’effet : soit la souris dort, soit elle ne dort pas.
Autre test : chez la souris, on mesure de l’activité
de type morphinique; injection de la morphine à
une certaine dose soit la queue de la souris est
dressée (réaction de Straub) soit elle ne l’est pas.
Réponse graduelle
On s’intéresse à intensité de l’effet en fonction de
la concentration :
Dose faible pas d’effet et plus on augmente la
concentration plus l’effet est important pour
arriver jusqu’à un effet maximum : saturation
A retenir :
Détermination de l’activité d’un agoniste : Effet (%)=f(concentration agoniste)
A savoir faire schématiquement.
Sur cette représentation on doit être capable
de placer le CE50
CE50 ou DE50: est la concentration de
l’agoniste nécessaire pour obtenir 50% de
l’effet maximal = puissance donc possible de
l’utiliser à dose faible
Plus elle est faible, plus le médicament est dit
puissant
20
II-Paramètres fonctionnels
21
II-Paramètres fonctionnels
Puissance= capacité à agir à
concentration (dose) faible

L2 Pharmacie - Pharmacologie N° 17
06/04/14 - P. Boulouard
Groupe 35 - Anais et Lucas
4
Théorie de l’occupation des récepteurs
L’occupation des récepteurs obéit à loi d’action de masse :
Affinité
Activité
intrinsèque
k1
[M]+[R]
[M-R]
Effet
pharmacologique
k2
[M]concentration en médicament
[R]concentration en récepteur
[M-R] concentration du complexe médicament récepteur
k1 constante cinétique d’association
Il existe un lien entre l’affinité et l’activité intrinsèque.
Réponse proportionnelle va être proportionnelle au % de récepteurs occupés par l’agoniste
Plus il y a de Rc liés plus la réponse est importante et vice versa
Réponse maximale = 100% d’occupation des récepteurs
Théorie de Clark (1933)
A+R AR avec K1= A x R/AR
Réponse = AR/Rt = AR/(R+AR)
Réponse = AR/Rt= A/A+K1
(Ne pas apprendre les formules, juste pour comprendre qu’il y a un lien entre la réponse et l’affinité)
Ariens (1954) = facteur de proportionnalité = αou activité intrinsèque
Réponse = α x A/(A+K1)

L2 Pharmacie - Pharmacologie N° 17
06/04/14 - P. Boulouard
Groupe 35 - Anais et Lucas
5
Activité intrinsèque d’un ligand agoniste
Réponse 100% : effet « complet » activité
intrinsèque α =1 AGONISTE ENTIER
Réponse partielle entre 0 et 100% : activité
intrinsèque 0<α<1 AGONISTE PARTIEL
Antagoniste neutre α = O pas de réponse
Agoniste inverse α < 0 réponse inverse à celle
d’un agoniste
(on doit être capable de refaire ou commenter ce
genre de schéma)
Agoniste entier est capable de produire un effet maximum tandis qu’un agoniste partiel n’est pas
capable d’atteindre cet effet.
Activité intrinsèque : capacité du ligand à produire un effet plu sou moins important en se liant au
récepteur
Notion de récepteur de réserve
Agoniste entier : effet max alors que tous les Rc
ne seraient pas occupés
Agoniste partielle même aux concentrations les
plus fortes aurait un effet moins important que
l’agoniste entier avec une occupation beaucoup
plus grande des Rc
A concentration égale entre un agoniste entier et
un agoniste partiel, l’un occupe partiellement les
Rc avec un effet supérieur et l’autre les occupe de
manière beaucoup plus importante avec un effet
partiel.
Mesure de courbe concentration pour différents agonistes
partiels comparativement à l’agoniste entier. On peut comparer
l’activité intrinsèque qui est l’effet en fonction de la
concentration au pourcentage d’occupation des récepteurs pour
différents types d’agonistes. (Juste à titre d’illustration)
L’activité intrinsèque permet de définir des agonistes
entiers, partiels et des antagonistes.
Notion de récepteur de réserve
23
24
 6
7
8
9
10
11
12
13
14
15
16
6
7
8
9
10
11
12
13
14
15
16
1
/
16
100%