LR RL →←+ - Page d`index

Pharmacologie_chap1.pdf
Chapitre1: Quantification de l'affinité, réponse, sélectivité
Évolution des concepts et modèles
Cible: lieu de fixation du médicament interagissant avec des récepteurs, des enzymes etc.
Affinité: interaction entre le médicament et sa cible.
Médicament + Cible Réponse
Réponse: somme des réponses des cellules, modification d’une fonction de l’organisme =
effet thérapeutique.
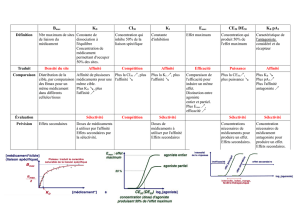
1. L’affinité et la loi d’action de masse
C'est un modèle mathématique qui sert de base pour quantifier l'affinité des médicaments.
La puissance de l'interaction physico-chimique entre le liguant et son récepteur correspond à
l'affinité réciproque des 2 partenaires. Cette affinité dépend de leur structure chimique
permettant l'établissement de liaisons non covalentes multiples (hydrophobes, ioniques, van
der walls…).
LRRL
k
→←+
1
L = ligand
R = récepteur
LR = complexe ligand récepteur
k
1
= constante cinétique d’association
k
-1
= constante cinétique de dissociation
L’analyse de la liaison des ligands aux récepteurs utilise un modèle mathématique simple dit
loi d’action de masse, équilibre dynamique entre les formes libres et associées du ligand et du
récepteur.
Ce modèle suppose que la liaison intervient lorsque le ligand et le récepteur se rencontrent en
fonction d’une simple diffusion, avec une orientation spatiale favorable des deux partenaires
et une énergie suffisante.
L’équilibre est atteint quand la vitesse d’association est égale à la vitesse de dissociation.
Vitesse d’association : L x R x k
1
nombre de phénomènes de liaison par unité de temps
Vitesse de dissociation : LR x k
-1
nombre de phénomènes de dissociation par unité de temps
A l’équilibre on a donc : L x R x k
1
= LR x k
-1
, soit :
D
K
k
k
LRRL ==
×
−
1
1
= constante de dissociation à l’équilibre, exprimée en concentration
molaire (M).
Elle sert à quantifier l'affinité du liguant pour le récepteur.
Selon la nomenclature actuelle, la constante K
D
est dénommée K
A
pour les agoniste et K
B
pour les antagonistes.
Strictement l’affinité est définie par la constante d’association à l’équilibre, K
a
pour un
agoniste, ou K
b
pour un antagoniste.
k
-1

Pharmacologie_chap1.pdf
D
ba Kk
k
KouKaffinité 1
1
1==⋅⋅= −
Toutefois, on utilise plus souvent K
A
ou K
B
que K
a
et K
b
car elles sont exprimées sous forme
molaire M.
Si le médoc A a un K
D
=10
-3
M et B a un K
D
= 10
-9
M. L'affinité est plus forte pour B.
Plus le K
D
est faible, plus l’affinité est élevée.
2. La réponse et la théorie d’occupation des récepteurs
L’affinité réciproque de L et R conduit à la formation du complexe LR.
La conséquence de la formation de LR est exprimée par différents termes relatifs à chaque
membre du complexe. Ainsi sont évoqués l’activité, ou effet de L, et son corollaire, la réponse
de R, termes équivalents en pratique.
La réponse de R peut être appréciée exceptionnellement au niveau des récepteurs eux-même
(ex : mesure d’un flux ionique pour un récepteur-canal)
, ou plus généralement à un niveau distal
(mesure
de la variation du taux d’un messager intracellulaire, réponse contractile ou sécrétoire de la cellule ou réponse
intégrée de l’organisme)
.
L’interprétation de cette réponse fait appel à la théorie d’occupation des récepteurs qui décrit
les relations quantitatives entre les concentrations de ligand et les réponses biologiques qui
résultent de l’interaction ligand-récepteurs. Cette théorie basée sur la loi d’action de masse
suppose que la réponse est directement proportionnelle au pourcentage de récepteurs occupés
par un agoniste et que la réponse maximale est obtenue avec 100% d’occupation des
récepteurs.
Donc plus on augmenterait la concentration plus il y aurait d'effet, or cette théorie est
fausse
.
La plupart du temps il faut stimuler un petit nombre de récepteurs pour avoir un effet max.
3. La sélectivité
« Toute substance est un poison et aucune n'est inoffensive. C'est simplement la dose qui fait
qu'une substance n'est pas toxique » Paracelse
Les effets indésirables sont du à la liaison d'un médicament à plusieurs cibles.
La sélectivité dépend de la dose. La sélectivité d’un ligand pour la cible R1 vis-à-vis de la
cible R2 correspond au rapport de son affinité pour R2 sur son affinité pour R1.
L’affinité étant l’inverse du K
D
, la sélectivité de L pour R1 vis-à-vis de R2 est égale au
rapport de K
DR2
/K
DR1
.
Pour être
sélectif
de R1 vis-à-vis de R2, ce rapport doit être
supérieur à 100
(2log)
On utilise alors le rapport des CE
50
ou DE
50
(dose pour laquelle l’effet correspond à 50% de l’effet max)
du
médicament L pour les effets E2 et E1. Ceci correspond à la notion de marge thérapeutique ou
différence entre les doses nécessaires à l’effet recherché et les doses entraînant un effet
secondaire voir toxique.
Aucun médicament n'est spécifique d'une cible.

Pharmacologie_chap1.pdf
4. Technique de liaison spécifique (binding)
Elles permettent de définir l'affinité de nouveaux ligands pour des sites de liaisons.
4.1. Incubation, séparation, mesure de L*R
Incubation : Ligand radioactif dans un tampon (pH, Temp précises) contenant les récepteurs
fixés sur une membrane. Les ligands se lient aux récepteurs avec un équilibre défini par la loi
d’action de masse.
Séparation par centrifugation ou filtration pour éliminer L* libre.
On peut déterminer le nombre de ligands liés aux récepteurs.
La membrane est mise dans un filtre à scintillation et on mesure la radioactivité, permettant de
quantifier L*R
Si on a une membrane quelconque le ligand peut se fixer ailleurs que sur le récepteur étudié.
La liaison du ligand à d’autres récepteurs/dans la membrane est appelée liaison no spécifique.
L’utilisation d’un ligand radiomarqué est toutefois longue et cher.
•
technique de saturation : on définit K
DL*
•
technique de déplacement : on définit K
i
L*
L*
L* L
*
Liaison non-spécifique
k
-1
* *
L R L R
+ ⇔
K
1

Pharmacologie_chap1.pdf
4.2. Technique de saturation
On
prend une série de tubes riches en récepteurs auxquels on ajoute des concentrations
croissantes de ligands radiomarqués (liaison totale).
Une seconde série est préparée parallèlement mais surchargée en ligand froid (liaison non
spécifique.
Le K
D
= concentration de L pour saturer
50% des récepteurs.
La représentation de Scatchard permet une détermination plus précise de K
D
et de
Bmax (la
densité de sites spécifiques).
Bound = L*R
Free = L*
* 1 max
*
*
D D
L R B
L R
L K K
−
= +
4.3. Technique de déplacement : Etude du déplacement d’un L* par un
ligand froid C (non radiomarqué)
Appelé aussi technique de compétition.
L’affinité du ligand non marqué pour son récepteur peut être appréciée en quantifiant son
pouvoir de déplacement d’un radioligand dont l’affinité (en pratique le K
D
) a été déterminée
préalablement par méthode de saturation.
Bound/free
Bmax
D
K
pente 1
−
=
)(* pMRL
D
K
Bmax

Pharmacologie_chap1.pdf
On met L* dans différents tubes en même quantité de manière à saturer les récepteurs.
Puis on ajoute un ligand inconnu C (compétiteur non marqué) à des concentrations
différentes.
Plus on augmente la concentration de C, plus C va déplacer L*. On laisse incuber puis on
mesure la radioactivité des L*R restant dans les tubes.
CI
50
: concentration qu’il faut pour déplacer 50% de L*.
Présence de deux équilibres :
-
entre L et R
-
entre C et R
CI
50
: valeur expérimentale
K
D
: paramètre
Avec
*
*
D
K L R
R
L
×
= et
i
C R
CR
K
×
= la valeur recherchée est K
i
50
*
1
i
D
CI
K
L
K
=+
La mesure faite est L*R en présence de C
La population totale des récepteurs est
*
Rt R CR L
= + +
soit
* ( )
L R Rt R CR
= − +
*.
*
(1 ) *
Di
l Rt
L R C
K L
K
=+ +
K
i
est une expression de K
D
pour une
expérience de compétition
100%
50%
R
L
*
50
CI
][
C
CR
RC
K
i
)(
×
=
R
L
RL
K
D
*
)*(
×
=
 6
7
8
9
6
7
8
9
1
/
9
100%