Récidive de la néphropathie cristalline après transplantation rénale : le déficit

Le Courrier de la Transplantation - Vol. XII - n° 4 - octobre-novembre-décembre 2012
186
Dossier thématique
Récidive de la néphropathie
initiale après transplantation
rénale sur le greffon
Résumé
Summary
»
Le déficit en adénine phosphoribosyltransférase (APRT) est
une maladie héréditaire rare, entraînant la formation de calculs
urinaires et, parfois, une insuffisance rénale sévère par précipitation
de cristaux dans le parenchyme rénal. Le déficit en APRT persiste
après une transplantation rénale, et la récidive de la néphropathie
cristalline peut mettre rapidement en péril la survie du greffon
rénal. La disponibilité de différents outils rend le diagnostic aisé
dès lors qu’il est évoqué. Il est crucial de ne pas méconnaître le
déficit en APRT chez le transplanté rénal afin de commencer le
plus tôt possible le traitement par allopurinol et de préserver la
fonction du greffon.
Mots-clés : APRT – Dihydroxyadénine – Cristaux – Transplantation.
Adenine phosphoribosyltransferase (APRT) deficiency is a
rare hereditary disease causing urolithiasis and, sometimes,
severe renal dysfunction secondary to precipitation of crystals
in renal parenchyma. APRT deficiency persists after renal
transplantation and crystalline nephropathy may recur
and lead to loss of graft function. Several diagnostic tools
available make the diagnosis fairly easy to confirm. It is of
crucial importance not to dismiss the possibility of APRT
deficiency in renal transplant recipients, in order to implement
allopurinol therapy early and preserve graft function.
Keywords: APRT – Dihydroxyadenine – Crystals –
Transplantation.
Récidive de la néphropathie cristalline
après transplantation rénale : le déficit
en adénine phosphoribosyltransférase
Recurrence of cristalline nephropathy after renal transplantation: APRT
deficiency
Guillaume Bollée*
U
ne grande variété de contextes cliniques
peuvent s’associer à l’accumulation dans
l’urine d’une substance au-delà de son seuil
de solubilité, entraînant la formation, l’agrégation et la
croissance de cristaux. Ce processus aboutit fréquem-
ment à la formation de calculs urinaires, dont la nature
est très variable en fonction du contexte métabolique
et clinique (oxalate ou phosphate de calcium, acide
urique, cystine, médicament, etc.) [1]. Cependant, la
survenue de calculs n’est pas la seule conséquence
pathologique de la cristallogenèse. Dans certaines
situations, les cristaux peuvent précipiter dans le paren-
chyme rénal, causant des lésions tubulo-interstitielles
sévères et, parfois même, une insuffisance rénale ter-
minale. Le déficit complet en adénine phosphoribo-
syltransférase (APRT) [2] est une maladie héréditaire
caractérisée par l’accumulation et la précipitation dans
l’urine de 2,8-dihydroxyadénine (DHA), pouvant causer
une néphropathie sévère évoluant vers l’insuffisance
rénale terminale. L’anomalie métabolique n’étant pas
corrigée par la transplantation rénale, la récidive de la
néphropathie cristalline sur le greffon est habituelle
en l’absence de traitement adéquat.
Dans le contexte de la transplantation rénale, il est
donc crucial de ne pas méconnaître cette maladie et
de prendre le plus tôt possible les mesures à même
d’assurer la meilleure survie rénale après la greffe. Le but
de cet article est de proposer une synthèse sur le déficit
en APRT, en insistant sur les aspects les plus importants
pour la prise en charge des patients en attente de greffe
ou transplantés rénaux.
Physiopathologie du déficit en APRT
L’APRT est une enzyme ubiquitaire, qui joue un rôle clé
dans le métabolisme des purines en catalysant la trans-
formation de l’adénine. Ainsi, l’adénine n’est présente
qu’à faible concentration dans le sang et l’urine dans les
conditions physiologiques. Chez les individus atteints
d’un déficit complet en APRT, l’adénine s’accumule et est
convertie par la xanthine oxydase en 8-hydroxyadénine
puis en 2,8-DHA (2). La DHA est éliminée dans l’urine, où
elle précipite en raison de sa faible solubilité, formant ainsi
des cristaux. Ces cristaux de DHA peuvent croître et donner
naissance à des calculs, ou bien précipiter dans le paren-
chyme rénal et causer une néphropathie cristalline (3, 4).
* Association pour
l’utilisation du rein
artificiel, Paris ;
Inserm U970, Paris.

Le Courrier de la Transplantation - Vol. XII - n° 4 - octobre-novembre-décembre 2012 187
En fonction du niveau d’activité enzymatique mesuré
in vitro dans les extraits cellulaires, 2 types de déficit en
APRT peuvent être distingués. Le déficit de type I, carac-
térisé par une activité enzymatique nulle in vitro, a été
décrit dans différents groupes ethniques et correspond
au déficit observé dans la population caucasienne (3).
Le déficit de type II, dû à une affinité réduite de l’APRT
pour le cofacteur 5-phosphoribosyl-1-pyrophosphate,
est caractérisé par une activité résiduelle in vitro et n’a
été décrit presque que dans la population japonaise (5).
En réalité, la distinction entre le type I et le type II est
sans conséquence clinique, l’enzyme APRT étant tota-
lement inactive dans les cellules en culture et proba-
blement in vivo, les individus atteints développant des
manifestations cliniques similaires (3, 5-7).
Le déficit en APRT est une maladie autosomique réces-
sive, et les individus atteints sont donc porteurs d’une
mutation dans les 2 copies du gène APRT. Dans le déficit
de type I, des mutations variées ont été identifiées,
regroupées sous le terme générique APRT*Q0 (3). Le
déficit de type II est lié à une mutation particulière
appelée APRT*J, et les sujets atteints ont le génotype
APRT*J/APRT*J ou, plus rarement, APRT*J/APRT*Q0 (5).
En pratique, il est utile de comprendre cette distinction
entre le type I et le type II pour bien interpréter le niveau
d’activité enzymatique mesuré sur les lysats cellulaires.
Ainsi, une activité APRT nulle signe un déficit complet,
mais une activité enzymatique diminuée peut corres-
pondre soit à un déficit partiel (sujet hétérozygote sain),
soit à un déficit complet in vivo (sujet homozygote
APRT*Q0 atteint). Cette dernière éventualité, fréquente
dans la population japonaise, semble exceptionnelle
dans les autres groupes ethniques.
Prévalence du déficit en APRT
La prévalence exacte du déficit en APRT est inconnue
et varie probablement selon les pays. Les cas rapportés
proviennent du Japon et, plus rarement, d’autres pays,
notamment l’Islande et la France (3, 6). Au Japon, la
fréquence du déficit complet en APRT a été estimée
à 1/27 000, et 1,2 % de la population seraient porteurs
d’une mutation à l’état hétérozygote (5). Dans les popu-
lations caucasiennes, la fréquence des hétérozygotes
a été estimée entre 0,4 et 1,2 % à partir de mesures de
l’activité APRT chez des sujets sains (8). Ainsi, la fré-
quence des mutations homozygotes serait de 1/50 000 à
1/100 000, ce qui est bien supérieur au petit nombre de
sujets identifiés et suggère que la maladie pourrait être
sous-diagnostiquée. Du fait de la fréquence particulière
de certaines mutations dans une population donnée,
la fréquence du déficit en APRT paraît particulièrement
élevée dans certains pays, comme le Japon, la France
et l’Islande (3, 5, 6). Cependant, une étude française a
permis d’identifier le déficit en APRT chez des individus
issus de groupes ethniques variés (3).
Présentation clinique du déficit en APRT
La présentation clinique et le mode de révélation du
déficit en APRT varient beaucoup. Les 2 types de mani-
festations de la maladie, à savoir la lithiase urinaire et la
néphropathie cristalline, peuvent être associés ou non,
et l’âge au début des symptômes est très variable. Dans
la cohorte française diagnostiquée à l’hôpital Necker,
l’âge de survenue du premier épisode de lithiase variait
entre 0,5 et 51 ans (moyenne : 19 ans) [3]. Une telle varia-
bilité a également été observée dans d’autres popula-
tions (5, 6). Il est donc possible de faire le diagnostic de
déficit en APRT aussi bien chez l’enfant que chez le sujet
âgé, parfois après 70 ans (3, 5, 6). Les calculs de DHA
sont pour la grande majorité radiotransparents, mais
peuvent être radio-opaques lorsqu’ils sont calcifiés (9).
En l’absence de diagnostic et de traitement précoce,
de nombreux patients développent une néphropathie
cristalline, due à la précipitation intratubulaire et inters-
titielle des cristaux de DHA. L’étude des cas français a
montré que 1/3 des patients présentaient une insuffi-
sance rénale chronique au moment du diagnostic (3).
La principale raison en est que le déficit en APRT est
souvent reconnu très tardivement, des années, voire
des décennies après l’apparition des symptômes (2, 3).
Parfois, la néphropathie cristalline survient chez des
patients n’ayant développé que quelques calculs, ce
qui rend le diagnostic difficile. L’évolution de la néphro-
pathie est le plus souvent subaiguë ou chronique, mais
une insuffisance rénale aiguë peut survenir, notamment
à l’occasion d’une déshydratation entraînant une oli-
gurie et la précipitation massive des cristaux.
Outils diagnostiques
Le diagnostic de déficit en APRT repose sur la mise en
évidence de DHA par l’étude des cristaux dans les urines
(parfois sur une biopsie rénale) ou par l’analyse d’un
calcul (2). L’analyse morphologique et constitutionnelle
par spectroscopie infrarouge d’un calcul, même ancien,
permet d’identifier la DHA dans tous les cas (10). L’étude
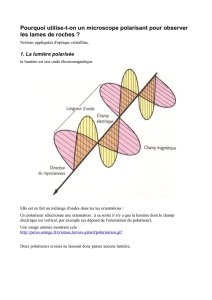
de la cristallurie en microscopie optique (1) a une sen-
sibilité et une spécificité proches de 100 % pour mettre
en évidence les cristaux de DHA (figure, A et B, p. 188),
qui ont, en lumière polarisée, un aspect caractéristique
en croix de Malte (figure, B, p. 188). L’analyse des cristaux
est complétée par la spectrophotométrie infrarouge
Récidive de la néphropathie cristalline

Le Courrier de la Transplantation - Vol. XII - n° 4 - octobre-novembre-décembre 2012
188
Dossier thématique
Récidive de la néphropathie
initiale après transplantation
rénale sur le greffon
pour confirmer l’identification de DHA. La biopsie rénale,
quoique théoriquement non nécessaire au diagnostic,
peut montrer des lésions tubulo-interstitielles sévères et
la précipitation de cristaux (figure, C). Il est essentiel de
savoir que l’aspect des cristaux visualisés sur la biopsie
rénale est généralement peu spécifique, et l’absence
d’aspect en croix de Malte n’écarte pas le diagnostic.
La présence de cristaux sur une biopsie rénale ne doit
jamais être négligée, et il est nécessaire de les carac-
tériser par l’étude de la cristallurie ou, directement sur
la biopsie rénale, par analyse infrarouge.
Bien que la présence de DHA soit pathognomonique
d’un déficit complet en APRT, le diagnostic peut être
confirmé par la mesure de l’activité enzymatique sur
lysats érythrocytaires à partir d’un simple prélèvement
sanguin, ce qui est relativement simple et représente
l’examen de référence. En France, cet examen peut être
réalisé dans le laboratoire de biochimie métabolique
de l’hôpital Necker à Paris (Dr Irène Ceballos-Picot). La
mesure de l’activité enzymatique est également utile
en première intention, lorsque l’analyse d’un calcul ou
de la cristallurie est impossible, chez le patient anurique
notamment. Dans le déficit de type I (population non
japonaise), l’activité APRT mesurée par cette technique
est nulle, alors qu’une activité résiduelle, correspondant
à 15 à 30 % des valeurs normales, est retrouvée dans
le type II (population japonaise) [5]. Très exceptionnel-
lement, il est toutefois possible d’observer un déficit
complet et une activité résiduelle sur les lysats érythro-
cytaires dans la population caucasienne, comme cela
a été rapporté dans une famille (11). Environ 90 % des
mutations en cause peuvent être mises en évidence par
le séquençage du gène APRT, mais l’étude génétique
n’est pas nécessaire au diagnostic (3).
Diagnostic du déficit en APRT
chez le patient en attente de greffe
ou le transplanté rénal
Après une transplantation rénale, le déficit enzymatique
persiste, et la récidive de la néphropathie cristalline sur
le greffon rénal est habituelle. Cela peut aboutir à la
perte irréversible de la fonction du greffon en quelques
semaines en l’absence de traitement adéquat (4, 9,
12-14).
Chez le patient en attente d’une greffe rénale et le trans-
planté, le déficit en APRT pose avant tout un problème
diagnostique. En effet, il n’est pas exceptionnel que
la maladie ne soit identifiée qu’après la récidive de la
néphropathie cristalline sur le greffon ; ce cas de figure
représentait même récemment 15 % des cas de déficit
en APRT diagnostiqués en France (3). Compte tenu de
la sévérité de la récidive après une transplantation,
il est essentiel d’évoquer la possibilité d’un déficit
en APRT chez tous les patients en attente de greffe
lorsque la nature de la néphropathie est incertaine et
de rechercher la notion d’épisodes lithiasiques, même
peu nombreux et anciens. Au moindre doute, l’étude de
la cristallurie ou la mesure de l’activité APRT sur un pré-
lèvement sanguin doit être réalisée. Malheureusement,
Figure. Cristallurie de 2,8-dihydroxyadénine (DHA) et néphropathie cristalline. A) Étude de la cristallurie en microscopie montrant
des cristaux de DHA arrondis, de couleur rouille (flèches). B) Aspect typique des cristaux de DHA dans l’urine avec une polarisation
caractéristique en croix de Malte (flèches). C) Étude en microscopie optique avec polarisation d’une biopsie de greffon rénal,
montrant la précipitation de cristaux dans les tubules (flèches) [trichrome de Masson, × 400]. L’analyse par spectrophotométrie
infrarouge des cristaux dans l’urine ou sur la biopsie rénale permet de confirmer leur nature.
(Remerciements au Dr L.H. Noël et au Dr M. Daudon, hôpital Necker, Paris, pour les clichés.)
A B C

Le Courrier de la Transplantation - Vol. XII - n° 4 - octobre-novembre-décembre 2012 189
il arrive que la transplantation soit réalisée sans que
la possibilité d’un déficit en APRT ait été écartée au
préalable. Le diagnostic doit alors impérativement être
évoqué devant la présence de cristaux sur une biopsie
du greffon rénal, même s’ils ne présentent pas de prime
abord l’aspect caractéristique en croix de Malte. Dans
une telle situation, l’étude soigneuse des cristaux en
microscopie à polarisation doit impérativement être
complétée par la spectrophotométrie infrarouge, qui
précisera la nature des cristaux. La recherche de cristaux
de DHA dans l’urine permet également de faire le dia-
gnostic dès les premiers jours après la greffe. Une fois
le diagnostic posé, une enquête familiale doit être réa-
lisée afin de rechercher d’autres sujets atteints, parfois
asymptomatiques (3, 6).
Traitement du déficit en APRT
Le traitement du déficit en APRT repose sur l’allopurinol,
qui inhibe la formation de la DHA en bloquant la xan-
thine oxydase, et sur des apports hydriques élevés (au
moins 2,5 l/j chez l’adulte) associés à un régime limité
en purines. L’alcalinisation des urines est inefficace et
n’est pas recommandée. La dose habituellement effi-
cace d’allopurinol est de 200 à 300 mg/j chez l’adulte
et de 5 à 10 mg/kg/j chez l’enfant. La posologie doit
être adaptée en cas d’insuffisance rénale. Ce traitement
permet habituellement d’obtenir une forte diminution,
voire la disparition des cristaux dans l’urine ainsi que
l’amélioration ou la stabilisation de la fonction rénale (3).
La surveillance de la cristallurie permet de s’assurer
de l’efficacité du traitement, et la persistance d’une
cristallurie abondante doit faire suspecter un défaut
d’observance ou une dose d’allopurinol insuffisante.
Bien que le nombre de patients suivis sur le long terme
soit limité, le traitement semble aussi efficace chez le
transplanté rénal et permet de prévenir la récidive de
la néphropathie cristalline sur le greffon. La bonne tolé-
rance et l’efficacité de l’allopurinol à long terme ont
été montrées chez l’enfant (15). En cas d’intolérance
à l’allopurinol, le fébuxostat, un autre inhibiteur de la
xanthine oxydase, pourrait représenter une alternative
thérapeutique, même si son utilisation n’a pas été rap-
portée dans le contexte du déficit en APRT.
Conclusion
Le déficit en APRT représente un cas de figure unique,
où une maladie évoluant fréquemment vers l’insuffi-
sance rénale terminale peut être traitée au moyen d’un
seul comprimé par jour. En effet, l’allopurinol permet
de bloquer la formation des cristaux de DHA à l’origine
des lithiases et de la néphropathie. Malheureusement,
le déficit en APRT est souvent diagnostiqué tardivement
et il arrive même que la maladie ne soit pas reconnue
avant le stade d’insuffisance rénale terminale. Compte
tenu du risque élevé de récidive de la néphropathie cris-
talline après la transplantation, il est essentiel de penser
à la possibilité d’un déficit en APRT chez les patients en
attente de greffe, lorsque la nature de la néphropathie
initiale est incertaine et qu’il y a des antécédents lithia-
siques, même peu nombreux. Tout retard diagnostique et
thérapeutique après la transplantation risque d’entraîner
la perte irrémédiable de la fonction du greffon rénal. ■
1. Daudon M, Jungers P. Clinical value of crystalluria and
quantitative morphoconstitutional analysis of urinary calculi.
Nephron Physiol 2004;98(2):31-6.
2. Bollée G, Daudon M, Knebelmann B, Ceballos-Picot I. Déficit
en adénine phosphoribosyltransférase (APRT). Actualités néphro-
logiques Jean Hamburger. Paris : Flammarion, 2010:99-108.
3. Bollée G, Dollinger C, Boutaud L et al. Phenotype and geno-
type characterization of adenine phosphoribosyltransferase
deficiency. J Am Soc Nephrol 2010;21(4):679-88.
4. Gagné ER, Deland E, Daudon M, Noël LH, Nawar T. Chronic
renal failure secondary to 2,8-dihydroxyadenine deposition: the
first report of recurrence in a kidney transplant. Am J Kidney
Dis 1994;24(1):104-7.
5. Kamatani N, Terai C, Kuroshima S, Nishioka K, Mikanagi
K. Genetic and clinical studies on 19 families with ade-
nine phosphoribosyltransferase deficiencies. Hum Genet
1987;75(2):163-8.
6. Edvardsson V, Palsson R, Olafsson I, Hjaltadottir G, Laxdal T.
Clinical features and genotype of adenine phosphoribosyltrans-
ferase deficiency in iceland. Am J Kidney Dis 2001;38(3):473-80.
7. Fujimori S, Akaoka I, Sakamoto K, Yamanaka H, Nishioka
K, Kamatani N. Common characteristics of mutant adenine
phosphoribosyltransferases from four separate Japanese
families with 2,8-dihydroxyadenine urolithiasis associated
with partial enzyme deficiencies. Hum Genet 1985;71(2):171-6.
8.
Johnson LA, Gordon RB, Emmerson BT. Adenine phosphori-
bosyltransferase: a simple spectrophotometric assay and the
incidence of mutation in the normal population. Biochem
Genet 1977;15(3-4):265-72.
9. Benedetto B, Madden R, Kurbanov A, Braden G, Freeman J,
Lipkowitz GS. Adenine phosphoribosyltransferase deficiency
and renal allograft dysfunction. Am J Kidney Dis 2001;37(5):E37.
10. Daudon M, Bader CA, Jungers P. Urinary calculi: review of
classification methods and correlations with etiology. Scanning
Microsc 1993;7(3):1081-104.
11. Deng L, Yang M, Fründ S et al. 2,8-Dihydroxyadenine
urolithiasis in a patient with considerable residual ade-
nine phosphoribosyltransferase activity in cell extracts but
with mutations in both copies of APRT. Mol Genet Metab
2001;72(3):260-4.
12. Eller P, Rosenkranz AR, Mark W, Theurl I, Laufer J, Lhotta
K. Four consecutive renal transplantations in a patient with
adenine phosphoribosyltransferase deficiency. Clin Nephrol
2004;61(3):217-21.
13. Glicklich D, Gruber HE, Matas AJ et al. 2,8-dihydroxyade-
nine urolithiasis: report of a case first diagnosed after renal
transplant. Q J Med 1988;68(258):785-93.
14.
Stratta P, Fogazzi GB, Canavese C et al. Decreased kidney
function and crystal deposition in the tubules after kidney
transplant. Am J Kidney Dis 2010;56(3):585-90.
15. Harambat J, Bollée G, Daudon M, Ceballos-Picot I, Bensman
A; APRT Study Group. Adenine phosphoribosyltransferase
deficiency in children. Pediatr Nephrol 2012;27(4):571-9.
Références bibliographiques
Récidive de la néphropathie cristalline
1
/
4
100%