Rôle de l ’ubiquitine ligase MARCH I dans l ’induction... des cellules dendritiques dans le diabète de type 1 ...

Université de Sherbrooke
Rôle de l’ubiquitine ligase MARCH I dans l’induction de la tolérance
des cellules dendritiques dans le diabète de type 1 (DT1) chez la souris
NOD
Par
Ahmed Benabdallah
Département de Pédiatrie, programme d’immunologie
Faculté de Médecine et des Sciences de la Santé
Mémoire présenté à la Faculté de Médecine et des Sciences de la Santé en vue de l’obtention du
grade de Maîtrise (M.Sc) en Immunologie
Novembre 2012

1+1 Library and Archives
Canada
Published Héritage
Branch
Bibliothèque et
Archives Canada
Direction du
Patrimoine de l'édition
395 Wellington Street
Ottawa ON K1A0N4
Canada
395, rue Wellington
Ottawa ON K1A 0N4
Canada
Your file Votre référence
ISB N: 978-0-494-94396-0
Our file Notre référence
ISBN: 978-0-494-94396-0
NOTICE:
The author has granted a non-
exclusive license allowing Library and
Archives Canada to reproduce,
publish, archive, preserve, conserve,
communicate to the public by
télécommunication or on the Internet,
loan, distrbute and sell theses
worldwide, for commercial or non-
commercial purposes, in microform,
paper, electronic and/or any other
formats.
AVIS:
L'auteur a accordé une licence non exclusive
permettant à la Bibliothèque et Archives
Canada de reproduire, publier, archiver,
sauvegarder, conserver, transmettre au public
par télécommunication ou par l'Internet, prêter,
distribuer et vendre des thèses partout dans le
monde, à des fins commerciales ou autres, sur
support microforme, papier, électronique et/ou
autres formats.
The author retains copyright
ownership and moral rights in this
thesis. Neither the thesis nor
substantial extracts from it may be
printed or otherwise reproduced
without the author's permission.
L'auteur conserve la propriété du droit d'auteur
et des droits moraux qui protégé cette thèse. Ni
la thèse ni des extraits substantiels de celle-ci
ne doivent être imprimés ou autrement
reproduits sans son autorisation.
In compliance with the Canadian
Privacy Act some supporting forms
may have been removed from this
thesis.
While these forms may be included
in the document page count, their
removal does not represent any loss
of content from the thesis.
Conformém ent à la loi canadienne sur la
protection de la vie privée, quelques
formulaires secondaires ont été enlevés de
cette thèse.
Bien que ces form ulaires aient inclus dans
la pagination, il n'y aura aucun contenu
manquant.
Canada

TABLE DES MATIÈRES
LISTE DES ABRÉVIATIONS
.............................................................................................................
iii
LISTE DES FIGURE ET DES TABLEAUX
.....................................................................................
iv
Figure 7: Schéma descriptif des plasmides vecteurs contenant les gènes d ’intérêts
..........
iv
Résumé
........................................................................................................................................................
v
CHAPITRE 1 : INTRODUCTION.
.......................................................................................................
1
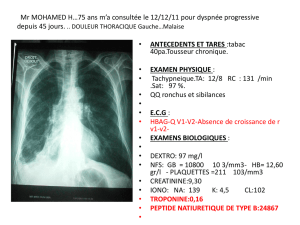
1. DIABÈTE DE TYPE I (DT1)
........................................................................................................
1
1.1. Définition
...................................................................................................................................
1
1.2. Épidémiologie et incidence du diabète
.................................................................................
2
1.2.1. Épidémiologie
...................................................................................................................
2
1.2.2. Incidence selon l’âge et le sexe
......................................................................................
2
1.3. Rôle des facteurs génétiques et environnementaux dans le développement du DT 1.4
1.3.1. Facteurs génétiques
..........................................................................................................
4
1.3.2. Facteurs environnem entaux
............................................................................................
5
1.4. Rôle des cellules immunitaires dans le développement et la progression du D T 1 6
1.4.1. Rôle des lymphocytes T
................................................................................................
7
1.4. 2. Rôle des lymphocytes iN K T
.......................................................................................
11
1.4. 3. Rôle des macrophages
.................................................................................................
11
1.4. 4. Rôle des cellules dendritiques
.....................................................................................
12
1.4.4.1. Cellules dendritiques (C D s)
..................................................................................
12
1.4.4.2. Ontogenèse et hétérogénéité des C D s
.................................................................
13
1.4.4.3. Caractéristiques des CDs
.......................................................................................
15
1.4.4.3.1 CDs immatures
..................................................................................................
15
1.4.4.3.2 CDs m atures
......................................................................................................
16
1.4.4.3.3 CDs semi-m atures
............................................................................................
18
1.4.4.4. Mécanisme d’ubiquitinylation impliqué dans la maturation des C D s 19
1.4.4.4.1. Mécanisme d’ubiquitinylation en général
................................................
19
1.4.(4.4.2 Protéine MARCH I dans son contexte historique
.....................................
22
1.4.4.4.3. MARCH I et le phénotype des CD s
...................
23
1.4.4.4.3. L’activation des lymphocytes T par les C D s
............................................
23
1.4.4.4.4. Rôle des CDs dans les conditions physiologiques
...................................
24

I.4.4.4.5. Cellules dendritiques et D T1
......................................................................
25
1.5. Modèles animaux du D T1
..................................................................................................
26
1.5.1. Modèles du diabète spontané
.......................................................................................
26
1.5.1.1. Souris non-obèse diabétique (N O D )
..................................................................
26
1.5.1.2. Rat BioBreeding
......................................................................................................
28
1.5.2. Modèles du diabète induit
.............................................................................................
29
HYPOTHÈSE ET OBJECTIFS
...........................................................................................................
30
CHAPITRE 2: MATÉRIELS ET M ÉTHODES
...............................................................................
31
2.1. Animaux
.......................................................................................................................................
31
2.2. Culture des cellules dendritiques
.............................................................................................
31
2.3 Analyse par cytométrie de flu x
.................................................................................................
32
2.4 Production de cytokines
.............................................................................................................
32
2.5 Production des lentivirus
............................................................................................................
33
2.5.1 Plasm ides
..............................................................................................................................
34
2.5.2 Cellules
.................................................................................................................
37
2.5.3 Production du lentivirus
.....................................................................................................
37
2.7 Transduction des cellules
...........................................................................................................
39
2.8 Détection des niveaux d'ARN messager par R T-PCR
..........................................................
39
2.9 Analyses statistiques
...................................................................................................................
40
CHAPITRE 3 : R ÉSU LTATS
.............................................................................................................
41
3.1 Phénotype des CDs générées en présence dT L -1 0
...............................................................
41
3.2 Profil des cytokines des IL-lO .C Ds
.........................................................................................
42
3.3 Niveau d’expression de MARCH I dans les IL-lO.CDs
......................................................
44
3.4 Effet de l’expression de MARCH I sur les CIITA-HEK 293
.............................................
45
3.5 Effet de la surexpression de MARCH I sur les C Ds
.............................................................
47
3.6 Effet de la surexpression des mutants À1-44 et A1-66 MARCH I sur les C D s
...............
49
CHAPITRE 4: DISCUSSION
..............................................................................................................
52
CONCLUSION ET PERSPECTIVES
................................................................................................
58
REMERCEMENTS
...............................................................................................................................
59
RÉFÉRENCES BIBLIOGRAPHIQUES
...........................................................................................
60
ii

LISTE DES ABRÉVIATIONS
Auto-Ac auto-anticorps
ADN acide désoxyribonucléique
ADNc acide désoxyribonucléique complémentaire
Ag antigènes
ARN acide ribonucléique messager
BMDC cellules dendritiques de la moelle osseuse
CCR motif C-C des récepteurs des chimiokines
CD classe de différentiation
CDs cellules dendritiques
CDsc cellules dendritiques conventionnelles
CMH complexe majeur d’histocompatibilité
CPA cellules présentatrices d’antigène
CSH cellules souches hématopoïétiques
CTL lymphocytes T cytotoxiques
CIITA cMH II transactivateur
DT1 diabète de type 1
Env enveloppe virale
ELIS A enzyme linked immunosorbent-assay
ELIS A enzyme linked immunosorbent-assay
E3 ligase E3
FBS sérum bovin fœtal
Gag groupe antigen
Foxp3 forkhead box protein 3
GIMAP5 GTPase immunity-associated protein family member5
GM-CSF granulocyte monocyte-colony stimulating factor
HLA antigènes du leucocytes humains
HEK293 humain embryonic kidney
iCDs CD immatures
idd diabète dépendant de l’insuline
IL interleukine
iNKT invariant natural killer T cell
LCMV chorioméningite lymphocytaire
LPS lipopolysaccharide
LT lymphocytes T
LTR long Terminal Repeat sequence
MARCH membrane-associated RING-CH
MIP-3a macrophage inflammatory protein-3a
MIR modulateur de la reconnaissance immunitaire
NOD non-obèses diabétiques
pCDs cellules dendritiques plasmacytoïdes
PCR polymerase chain reaction
Pol polymérases
RT-PCR reverse transcriptase-polymerase chain reaction
Tat transactivator of transcription
TLR toll-like receptor
TNF tumor necrosis factor
VIH virus de l’Immunodéficience Humaine
iii
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
1
/
78
100%