R é u n i o n Réunion OXYGÈNE, VAISSEAUX ET CANCER

Réunion
Réunion
35
OXYGÈNE, VAISSEAUX ET CANCER
J. Pouysségur (Institut de signalisation, biologie et développe-
ment du cancer, Centre A. Lacassagne, Nice)
Le soleil est notre source de vie. Il circule dans nos artères
sous forme d’oxygène. Le soleil transforme le gaz carbonique
en une flore luxuriante. Celle-ci excrète l’oxygène, qui devient
source de vie pour la faune animale. Ainsi, l’astre solaire marie
l’homme au végétal. Alors que nos artères véhiculent tous les
nutriments indispensables à notre croissance et survie, ce sont
les variations de teneur en oxygène d’un organe qui "sculptent”
son réseau vasculaire. Grâce à des détecteurs d’oxygène sophis-
tiqués – proline et asparagine hydroxylases, qui contrôlent
l’activité transcriptionnelle du facteur HIF –, les organes densi-
fient ou élaguent le réseau vasculaire en fonction de la teneur
en oxygène du tissu. Ce phénomène naturel est exploité par les
tumeurs. Leur croissance épuise rapidement l’oxygène local, un
signal déclenchant la vascularisation (angiogenèse tumorale),
le développement de la tumeur et souvent la dissémination des
cellules. Les cancers du côlon et du rein bénéficient depuis peu
de traitements anti-angiogéniques. Nous essayerons d’apporter
lumière et air frais dans ce domaine de la biologie (1).O
RÉFÉRENCE BIBLIOGRAPHIQUE
1. Pouysségur J, Dayan F, Mazure NM. Hypoxia signalling in cancer and approa-
ches to enforce tumour regression. Nature 2006;441:437-43.
LES ANTIANGIOGÉNIQUES
DANS LES CANCERS DIGESTIFS
D. Malka (Institut Gustave-Roussy, Villejuif)
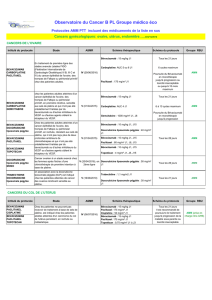
Après avoir rappelé le contexte actuel de la prise en charge
du cancer colorectal (CCR), D. Malka présente les principaux
antiangiogéniques disponibles selon leur mécanisme d’action
et le niveau de leur développement en recherche clinique.
Dans le CCR métastatique, la première molécule antiangiogénique
(et la seule à ce jour) à avoir obtenu une autorisation de mise sur le
marché (AMM) en association avec de la chimiothérapie en première
ligne thérapeutique est Avastin© (bévacizumab) [tableau I].
L’étude d’enregistrement (1) a randomisé 815 patients présentant
un CCR métastatique entre un traitement par IFL (irinotécan,
5-fluoro-uracile [5-FU], acide folinique [AF]) + placebo versus
IFL + bévacizumab (B) [à la dose de 5 mg/kg/14 jours]. L’aveugle
a été levé à la progression, et les patients sous B pouvaient conti-
nuer à recevoir l’anti-VEGF en combinaison avec un traitement
de seconde ligne. L’objectif principal de l’étude était la survie
globale. Comme prévu selon le protocole, un troisième bras
(5-FU/AF/B) a été interrompu après vérification de la faisabilité
de la combinaison IFL + B.
Tableau I.
Antiangiogéniques en cours de développement.
Agents Firme Type (cibles) Développement
Bévacizumab* Roche/Genentech ACM (VEGF-A) Commercialisé
VEGF-Trap Regeneron VEGFR1 soluble Phase I/II
Antagonistes VEGFR
Bay 43-9006
(sorafenib) Bayer/Onyx IRTK**, IRAFK Phase III
Valatinib
(PTK787/ZK222584)*
Novartis/
Schering Plough
IRTK** (VEGFR1/2,
PDGFR, c-KIT) Phase III
Angiozyme Sirna Ribozyme
(ARNm VEGFR1/2) Phase II
ZD6474 AstraZeneca IRTK**
(VEGFR1/2, EGFR) Phase II
AG013676 Pzer IRTK** Phase II
SU11248 (sunitinib) Pzer IRTK**
(VEGFR, PDGFR, c-KIT) Phase I/II
GW786034 GlaxoSmithKline IRTK (VEGFR) Phase I/II
IMC-IC11 ImClone ACM (VEGFR2) Phase I
ACM : anticorps monoclonal, IRTK : inhibiteur oral de récepteur(s) à tyrosine kinase,
IRAFK : inhibiteur de RAF kinase.
* Résultats en phase III rapportés au cours du CCR.
** Inhibent divers RTK, dont VEGFR1, VEGFR2, PDGFR, c-kit ou Flt-3.
Les patients randomisés dans le bras IFL + B ont eu significative-
ment plus de réponses que ceux du bras IFL + placebo (44,9 %
versus 34,7 % ; p = 0,003) ; la survie sans progression a été significa-
tivement améliorée (10,6 mois versus 6,2 mois ; p < 0,00001), ainsi
que la survie globale (20,3 mois versus 15,6 mois ; p = 0,00003).
Un traitement de deuxième ligne a été administré à 53 % des
patients du groupe IFL + placebo et à 61 % de ceux du groupe
IFL + B. Pour les malades traités en seconde ligne par oxaliplatine,
le bénéfice en termes de survie globale persistait dans le groupe
initialement traité par IFL + B (25,1 mois) par rapport à celui
ayant reçu IFL + placebo (22,2 mois).
Compte-rendu des Premières Rencontres francophones
sur l’angiogenèse tumorale – Avignon, le 24 juin 2006
D'après les présentations de J. Pouysségur, D. Malka, G. Pagès, G. Tobelem, J.P. Delord, F. Tranquart, X. Pivot, J. Balosso
recueillies par L. Aimard (Clinique Clairval, Marseille).
La Lettre du Cancérologue - Vol. XVI - n° 1-2 - janvier-février 2007
LK1-BAG_0207.indd 35 26/02/07 12:19:47

Réunion
Réunion
36
Une étude rétrospective a analysé la survie globale observée en
tenant compte des critères de Köhne et al. (statut de perfor-
mance, nombre de sites tumoraux, phosphatases alcalines,
globules blancs) [2].
Ainsi, la survie globale était significativement améliorée si le
traitement administré était IFL + B (quel que soit le groupe
pronostique) et si le risque était faible (quel que soit le schéma
utilisé). Le bénéfice lié au B était observé même chez les patients
de mauvais pronostic (tableau II).
Dans l’étude de Hurwitz et al., en termes de tolérance, au total,
85 % des patients du bras B et 74 % de ceux du bras placebo ont
présenté une toxicité de grade 3-4 (p < 0,01). Les pourcentages
de décès toxiques et de décès dans les 60 premiers jours ne
différaient pas d’un groupe à l’autre (2,5 % versus 2,8 % ; 3 %
versus 4,9 %). Les toxicités principales ont été la diarrhée de
grade 3-4 (33 % versus 25 %) et la neutropénie de grade 3-4 (37 %
versus 31 %). Une attention toute particulière a été portée aux
événements vasculaires : une hypertension artérielle (HTA) a
été plus fréquemment observée sous B (22,4 % versus 8,3 %),
mais les événements thrombo-emboliques (19,3 % versus 16,1 %)
ou hémorragiques (3,1 % versus 2,5 %) n’ont pas différé d’un
groupe à l’autre. Six patients (1,5 %) du bras B ont présenté une
perforation digestive (1).
En termes de tolérance, les observatoires relatifs à l’utilisation
de B dans le cadre des AMM délivrées par la Food and Drug
Administration et l’European Medicines Agency (EMEA) [études
BRiTE et First BEAT] montrent des résultats comparables à ceux
observés dans les essais randomisés (tableau III). Ainsi, aucun
nouvel événement indésirable n’a été détecté ; les perforations et
les hémorragies digestives sont tumorales dans environ 50 % des
cas, avec un risque accru si le cancer primitif n’est pas réséqué
(fatales : 2,2 % versus 0,3 %) [3-6].
En ce qui concerne les complications de la chirurgie faite avant
ou après la mise sous B, l’analyse issue des données de l’étude
pivotale a été publiée (7). Le tableau IV reprend les résultats.
Tableau IV. Combinaison chimiothérapie (CT) + bévacizumab (B) :
morbidité opératoire.
Chimiothérapie seule
(n = 516)
Chimiothérapie + B
(n = 616)
Chirurgie avant utilisation de B 194 230
Complications 1 (0,5 %) 3 (1,3 %)
Chirurgie
au cours de l’utilisation du B 29 75
Complications 1 (3,4 %) 10 (13,3 %)
Pour une chirurgie plus spécifique telle que la chirurgie hépa-
tique, plusieurs points sont abordés, mais peu de réponses sont
actuellement disponibles.
Ainsi :
il n’y a pas de données sur la régénération hépatique et anti-
VEGF ;
l’impact clinique de la toxicité de la chimiothérapie est une
notion controversée ;
les anti-VEGF inhibent la cicatrisation ;
la demi-vie du B est longue (17-21 jours) ;
le délai proposé entre la dernière administration de B et une
chirurgie est de 8 semaines.
Lors du congrès de l’ASCO 2006, deux équipes ont présenté les
résultats de l’utilisation de B dans le contexte de la chirurgie
des métastases hépatiques (tableau V) [8-9].
Tableau V. Combinaison chimiothérapie (CT) + bévacizumab (B) :
morbidité après hépatectomie.
Étude [8]
(n = 19 évaluables/
34 opérés de l’étude)
Étude [9]
(n = 43 opérés/
1927 pts de l’étude)
Type d’étude Phase II First BEAT
Population Patients potentiellement
opérables Patients non opérables
Chimiothérapie XELOX 100 % Oxaliplatine (60 %),
CPT11 (37 %)
Délai B/chirurgie 5 semaines 8 semaines
Complications
postopératoires
5%
Fuite biliaire
Abcès de paroi
6%
liés au B
(33 % en tout)
Tableau II. Survie globale selon la classe de risque pronostique
(d’après [2]).
Survie globale médiane (mois)
Risque IFL + placebo
(n = 401)
IFL + B
(n = 411)
faible (n = 233) 19,8 25,7
moyen (n = 493) 15,3 20,1
élevé (n = 86) 10,4 14,3
Tableau III. Combinaison chimiothérapie (CT) + bévacizumab (B) : toxicité.
Eets
indésirables (%)
AVF2107g
(États-Unis)
[1]
BRiTE
(États-Unis)
[3]
First BEAT
(Europe, etc.)
[4-6]
IFL + B
(n = 393)
Chimiothérapie
(CT) + B
(n = 1968)
CT + B
(n = 1915)
Thrombo-embolies
artérielles 3,3 2,1 0,6
Hémorragies
grade 3-4 3,1 1,9 3,6
Perforations
digestives 1,5 1,7 1,5
La Lettre du Cancérologue - Vol. XVI - n° 1-2 - janvier-février 2007
LK1-BAG_0207.indd 36 26/02/07 12:19:48

Réunion
Réunion
37
La conclusion retenue par les différents auteurs est que la
chirurgie hépatique (métastases résécables d’emblée ou secon-
dairement) de patients traités par B paraît possible dans de
bonnes conditions de sécurité, moyennant un délai d’au moins
5 semaines après la dernière administration de B.
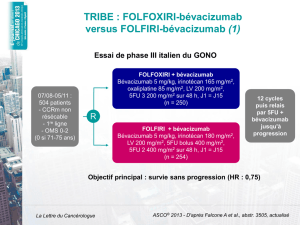
Toujours en première ligne de traitement du CCR métastatique,
l’étude TREE-1 compare trois différents schémas d’administration
de fluoropyrimidines et d’oxaliplatine (FOLFOX ou 5-FU
bolus ou capécitabine). Puis l’étude de phase II TREE-2 a suivi,
en gardant les mêmes combinaisons de chimiothérapie (doses
réduites de capécitabine), mais en associant B dans chaque
bras (10).
Même si l’on ne peut effectuer de comparaison méthodolo-
giquement correcte entre ces deux études, on peut retenir que
(tableau VI) :
les trois schémas d’administration sont efficaces, avec un taux
de réponse plus faible et une survie sans progression inférieure
pour l’association avec du 5-FU en bolus (bFCL) ;
l’adjonction de B à chacun des trois schémas augmente le
taux de réponse, le temps jusqu’à progression et la survie globale
sans augmenter la toxicité.
En deuxième ligne de traitement du CCR métastatique, une
étude de phase III randomisée a comparé FOLFOX 4, B mono-
thérapie haute dose (10 mg/kg/15 j) à l’association des deux dans
le CCR avancé ou métastatique prétraité par une chimiothérapie
associant 5-FU et CPT11 (11).
L’objectif principal (amélioration de la survie globale de 7 à
9,8 mois [+ 40 %] par l’adjonction de B au FOLFOX 4) et les
objectifs secondaires (taux de réponse, survie sans progression,
tolérance) ont été atteints, avec une amélioration significative du
taux de réponse (21,8 % versus 9,2 %), de la survie sans maladie
(7,2 mois versus 4,8 mois ; p < 0,001) et de la survie globale
(12,9 mois versus 10,8 mois) dans le bras FOLFOX + B.
Cette étude démontre la supériorité de l’association FOLFOX + B
sur FOLFOX dans le traitement des CCR avancés prétraités
par irinotécan.
Dans cette indication, B en monothérapie (bras thérapeutique
prématurément fermé) n’est pas efficace.
Du point de vue de la tolérance, on observait une augmentation
du risque d’HTA, de saignement et de neuropathie dans le bras
FOLFOX + B. Le taux de perforation digestive a été comparable
à celui observé dans les autres études (1 % bras FOLFOX + B
versus 1,3 % bras B), et il n’y a pas d’augmentation significative
du risque de thrombose veineuse ou artérielle.
Une autre étude de phase II randomisée a comparé les associa-
tions CBI (B + cétuximab [C] + CPT11) et CB chez des patients
atteints d’un CCR métastatique réfractaire à l’irinotécan (12).
La recherche de l’expression d’EGFR n’était pas un prérequis
pour l’inclusion.
Les patients ont été traités, en médiane, en troisième ligne après
progression sous CPT11 dans 100 % des cas et sous oxaliplatine
dans 89 % des cas.
Une augmentation du taux de réponse objective (37 % versus
20 %) et de la survie médiane sans progression (7,9 mois
versus 5,6 mois) a été observée dans le bras CBI par rapport
au bras CB.
La toxicité liée à B et C a été comparable entre les deux bras ;
les autres toxicités ont été plus fréquentes dans le bras CBI
(neutropénie grade 3-4 [22 % versus 0 %], diarrhée grade 3-4
[24 % versus 0 %], asthénie grade 3 [10 % versus 0 %]).
Une comparaison historique avec des patients traités par C seul
ou en association avec CPT11 suggère que l’ajout de B améliore
l’efficacité (chez des patients n’ayant jamais reçu de B auparavant).
Les études cliniques en cours testant l’utilisation des antiangio-
géniques dans le traitement adjuvant du CCR sont résumées
dans le tableau VII.
Tableau VII.
Combinaisons chimiothérapie (CT) + bévacizumab (B) :
essais en cours en adjuvant.
Essai Schémas Eectif
AVANT BO17920
FOLFOX 4
FOLFOX 4 + B
XELOX + B
3 450
NSABP C-08 mFOLFOX 6
mFOLFOX 6 + B 2 600
QUASAR 2
Capécitabine
CAPIRI
CAPIRI + B
3 750
ECOG 5205 FOLFOX 6
FOLFOX 6 + B 3 125
Tableau VI.
Associations uoropyrimidine + oxaliplatine : apport du bévacizumab (d’après [11]).
TREE-1 TREE-2
mFOLFOX bFOL CAPOX mFOLFOX + B bFOL + B CAPOX + B
Taux de réponse (%) 43 22 35 53 41 48
Temps jusqu'à progression (mois)
8,7 6,9 5,9 9,9 8,3 10,3
Survie globale (mois) 19,2 17,9 17,2 26,0 20,7 27,0
Toxicité grade 3-4 (%) 59 36 67 59 51 56
La Lettre du Cancérologue - Vol. XVI - n° 1-2 - janvier-février 2007
LK1-BAG_0207.indd 37 26/02/07 12:19:48

CELLULE
CANCÉREUSE
CELLULE
ENDOTHÉLIALE
Survie
Croissance
VEGF
PTEN
Ras
PI3-K/Akt
p42/p44 MAPK
p42/p44
MAPK
R2
Akt
PI3-K
R1
Figure 1.
Activation combinée des voies de signalisation PI3
kinase et MAP kinases/ERK dans des cellules cancéreuses et les
cellules endothéliales entraînant leur croissance et leur survie.
Un acteur paracrine majeur, le VEGF, produit par les cellules can-
céreuses, provoque un phénomène d'angiogenèse qui amplie
le développement de la tumeur (d'après [3]).
Oncogenes
Growth factors
HIF-1α
stabilisation
Stress kinases
JNK/p38MAPK
c Jun
c Fos
AP-2 mRNA
stabilisation
HIF-1
Hypoxia
Sp1 VEGF
p42/p44 MAPK
+
+
+
+
αβ
Figure 2.
Représentation schématique de la régulation de l'ex-
pression du VEGF (d'après [3]). Les aspects majeurs de cette
régulation sont discutés dans le texte et dans les articles 1 à 4.
Réunion
Réunion
38
Enfin, dans le domaine des cancers digestifs non colorectaux
tels que le carcinome hépatocellulaire, le cancer du pancréas
ou de l’estomac, aucun essai de phase III n’est encore disponible
avec les anti-VEGF. O
RÉFÉRENCES BIBLIOGRAPHIQUES
1. Hurwitz et al. New Engl J Med 2004.
2. Kabbinavar et al. ASCO 2006, abstract 3539.
7. Scappaticci et al. J Surg Oncol 2005;91:173.
11. Giantonio. ASCO 2005, abstract 2.
12. Saltz et al. ASCO GI 2005, abstract 169b.
DÉTERMINANTS CIBLES : VEGF, VEGF-R.
PEUT-ON AIDER LE CLINICIEN DANS LA DÉCISION
D’UN TRAITEMENT PAR ANTIANGIOGÉNIQUE ?
G. Pagès (université de Nice Soa Antipolis/Institut de signalisa-
tion Biologie et développement du cancer)
Deux voies majeures sont mutées au cours de la carcinogenèse
humaine : Ras-extracellular signal regulated kinases (ERK) et
la phosphatidylinositol-3OH kinase (PI[3]K)-AKT. Elles sont
activées par de nombreux polypeptides facteurs de croissance,
hormones ou protéines de la matrice extra-cellulaire. Les voies
de signalisation Ras/Raf/MEK/ERK et PI(3)K-AKT interviennent
dans l’expression du facteur ubiquiste que représente le VEGF-A,
facteur clé de la vascularisation et de l’angiogenèse. Le but de
cette présentation est de montrer l’impact de la voie ERK sur
l’expression de VEGF-A et sur le développement tumoral.
Voie de signalisation des MAP kinases/ERK et VEGF-A
Les MAP kinases/ERK appartiennent à une grande famille
de sérine/thréonine kinases activées par de multiples signaux
extra-cellulaires, comme des facteurs de croissance, hormones,
lymphokines, facteurs de stress, composants de la matrice extra-
cellulaire. Il existe deux isoformes p42/p44 MAP kinases, égale-
ment appelées ERK1/ERK2. Elles interviennent de façon majeure
dans l’expression de VEGF-A, en particulier via l’augmentation
de la transcription de son gène par l’intermédiaire de la phos-
phorylation des facteurs de transcription HIF-1 (1) et Sp1 (2)
[figures 1 et 2].
Les cascades de signalisation des MAP kinases/ERK ainsi que celle
des kinases du stress p38 et JNK interviennent également dans
les processus de stabilisation de l’ARNm du VEGF-A (4).
La voie de signalisation Raf/MEK/ERK, en plus de son impor-
tance pour la production du VEGF-A par les cellules tumorales,
est également stimulée à la suite de l’activation de nombreux
récepteurs présents sur les cellules endothéliales, cibles du
VEGF-A, mais aussi sur les cellules tumorales (récepteurs du
VEGF, de l’EGF et du PDGF). Cette voie de signalisation se
révèle être une cible thérapeutique intéressante, son inactiva-
tion pouvant à la fois bloquer les mécanismes de prolifération
cellulaire et d’angiogenèse et promouvoir l’apoptose.
VEGF et ecacité thérapeutique
Les niveaux de VEGF-A intratumoraux ont été quantifiés par la
technique ELISA sur 54 échantillons de tumeurs ORL humaines
diagnostiquées au centre Antoine-Lacassagne de Nice. Le VEGF
s’est révélé être un facteur pronostique indépendant de survie
sans récidive, mais aussi de survie globale. De même, les niveaux
plasmatiques de VEGF-A sont plus élevés chez des patients
atteints de leucémie myéloïde chronique que chez des personnes
saines. L’efficacité de l’imatinib, une thérapie ciblée sur la chimère
Bcr/Abl, responsable de la maladie, peut être suivie par mesure
des taux de VEGF circulants, ceux-ci devenant normaux chez
des patients en rémission cytogénétique (5). Sur des modèles
cellulaires de cancer du sein ou de la prostate métastatique, il
est relativement aisé d’obtenir des cellules résistantes au docé-
taxel, un traitement couramment utilisé en clinique. Nous avons
démontré que le docétaxel activait le promoteur du VEGF-A
et induisait des accumulations intracellulaires de VEGF-A. Ces
résultats mettent en évidence l’un des mécanismes potentiels
de résistances. Ils montrent également l’intérêt de l’association
La Lettre du Cancérologue - Vol. XVI - n° 1-2 - janvier-février 2007
LK1-BAG_0207.indd 38 26/02/07 12:19:49

HIF - α
HIF - β
PDCF
RPLG2P13K
Cellule
endothéliale,
péricyte
ou cellule
tumorale
Ral
MEX
ERK
EGF
VEGF
pVHLmTOR
BAY 43 -
SU011248
Getinib, cétuximab
erlotinib, ABX-EGF
BAY 43 - 9006
- 5132
CI - 1040
BAY 43 -
PTK/2K
SU011248
Bévacizumab
CCI-779
Figure 3.
Sites d'actions d'agents thérapeutiques ciblés dans le
traitement du carcinome rénal.
Réunion
Réunion
39
docétaxel + bévacizumab, un anticorps monoclonal ciblant le
VEGF-A. De même, dans le cadre du cancer du sein métastatique
surexprimant le récepteur HER-2, l’utilisation du trastuzumab,
anticorps monoclonal ciblant ce récepteur, inhibe la voie de
signalisation Raf/MEK/ERK. Le gefitinib, un inhibiteur de tyro-
sine kinase ciblant le récepteur de l’EGF, a également montré une
efficacité importante dans le cancer du poumon ou les cancers
ORL et ce, également par inhibition de la voie Raf/MEK/ERK.
L’inhibition de cette voie de signalisation apparaît donc comme
une cible thérapeutique majeure. Des essais précliniques chez la
souris sont en cours pour évaluer l’impact d’inhibiteurs molécu-
laires ou pharmacologiques spécifiques des membres de cette
voie de signalisation sur le développement de cancers du sein,
du pancréas ou du côlon et de mélanomes.
Perspectives
Une meilleure appréciation de la réceptivité tumorale s’avère
nécessaire pour mieux adapter les différentes thérapeutiques
antiangiogéniques. Pour l’angiogenèse, deux molécules présen-
tent un intérêt thérapeutique évident : le bévacizumab, qui cible
le VEGF-A, et le sunitinib, qui cible plutôt ses récepteurs (VEGF-
R1 et 2). Il est important de souligner que, lorsque l’expression de
VEGF-A est diminuée, d’autres facteurs angiogéniques prennent
le relai pour continuer à promouvoir une angiogenèse efficace.
Un de ces facteurs “relai” est sans conteste l’interleukine 8, un
facteur de croissance des cellules endothéliales aussi efficace
que le VEGF-A. Ce relai angiogénique pourrait expliquer une
efficacité moins importante qu’attendue des anti-VEGF-A. Un
autre aspect prenant un essor important pour la dissémination
métastatique s’avère être les mécanismes de lymphangiogenèse.
Des facteurs apparentés au VEGF-A sont des acteurs importants
de ce mécanisme. Il s’agit des VEGF-C et VEGF-D et de leur
récepteur, le VEGF-R3. Cependant, aucune molécule ciblant
ces facteurs ou leur récepteur n’est disponible actuellement.
Il est intéressant de noter que la liaison des VEGF-C ou des
VEGF-D au VEGF-R3 active la voie de signalisation Ras/Raf/
MEK/ERK. La multiplicité des voies de signalisation mises en
jeu lors des processus de tumorisation mais aussi l’apparition de
phénomènes d’adaptation lors de traitements faisant intervenir
un seul composé (figure 3) impliquent que les thérapeutiques
combinées vont devenir la règle. Cependant, leur application en
clinique va nécessiter de nombreux travaux pour mieux évaluer
les différentes modalités et périodicités d’administration. O
RÉFÉRENCES BIBLIOGRAPHIQUES
1. Richard DE, Berra E, Gothie E, Roux D, Pouysségur J. P42/p44 mitogen-
activated protein kinases phosphorylate hypoxia-inducible factor 1alpha (HIF-
1alpha) and enhance the transcriptional activity of HIF-1. J Biol Chem 1999;274
(46):32631-7.
2. Milanini-Mongiat J, Pouysségur J, Pagès G. Identification of two Sp1 phospho-
rylation sites for p42/p44 mitogen-activated protein kinases: their implication
in vascular endothelial growth factor gene transcription. J Biol Chem 2002;
277(23):20631-9.
3. Berra E, Pagès G, Pouysségur J. MAP kinases and hypoxia in the control of
VEGF expression. Cancer Metastasis Rev 2000;19:139-45.
4. Pagès G, Berra E, Milanini-Mongiat J et al. Stress-activated protein kinases
(JNK and p38/HOG) are essential for vascular endothelial growth factor mRNA
stability. J Biol Chem 2000;275(34):26484-91.
5. Legros L, Bourcier C, Jacquel A et al. Imatinib mesylate (STI571) decreases
the vascular endothelial growth factor plasma concentration in patients with
chronic myeloid leukemia. Blood 2004;104(2):495-501.
6. Stadler WM. Targeted agents for the treatment of advanced renal cell carci-
noma. Cancer 2005;104:2323-33.
BIOLOGIE DE L’ANGIOGENÈSE
G. Tobelem (Institut des vaisseaux et du sang, Paris)
Historique
Le champ de recherche de l’angiogenèse démarre en 1971 avec la
publication de “Tumor angiogenesis and therapeutic implications”
de Judah Folkman, dans le New England Journal of Medicine.
Cette hypothèse postulait que l’angiogenèse était un phénomène
incontournable pour la progression tumorale, mais également que
les antiangiogènes pourraient entraîner sa régression. J. Folkman
développe alors le concept de switch angiogénique.
Switch angiogénique : les années 1970
La croissance normale et néoplasique est sous la dépendance
de l’angiogenèse.
Des cancers in situ non angiogéniques pourraient chez
l’homme survenir dans plusieurs organes, mais un tout petit
nombre (1 sur 600) basculerait vers un phénotype angiogénique,
donnant ainsi une tumeur détectable.
Des facteurs antiangiogènes naturels contrebalancent les facteurs
angiogènes, mais cet équilibre se rompt dans la maladie cancéreuse
alors qu’il persiste au cours de la cicatrisation ou de l’ovulation.
L’endothélium vasculaire recruté par une tumeur est une cible
thérapeutique secondaire intéressante puisque génétiquement
stable et moins susceptible de résistance (1).
La Lettre du Cancérologue - Vol. XVI - n° 1-2 - janvier-février 2007
LK1-BAG_0207.indd 39 26/02/07 12:19:49
 6
7
8
9
10
11
6
7
8
9
10
11
1
/
11
100%