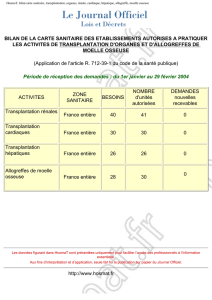
THESE DOCTORAT DE L’UNIVERSITÉ DE TOULOUSE En vue de l'obtention du

THESE
THESE
En vue de l'obtention du
DOCTORAT DE L’UNIVERSITÉ DE TOULOUSE
DOCTORAT DE L’UNIVERSITÉ DE TOULOUSE
Délivré par l'Université Toulouse III - Paul Sabatier
Discipline ou spécialité : Physiopathologie
JURY
Dr. Nassim Kamar, Maitre de Conférences-Practicien hospitalier (CHU Toulouse) Directeur de thèse
Pr. Robert Salvayre, Professeur-Practicien hospitalier (CHU Toulouse) Directeur de thèse
Pr. Lionel Rostaing, Professeur-Practicien hospitalier (CHU Toulouse) Examinateur
Pr. Pierre Marquet, Professeur-Practicien hospitalier (CHU Limoges) Rapporteur
Pr. Christophe Mariat, Professeur-Practicien hospitalier (CHU Saint-Etienne) Rapporteur
Ecole doctorale : Ècole doctorale biologie santé et biotechnologies
Unité de recherche : I2MR/INSERM U858-Èquipe 10
Directeur(s) de Thèse : Dr Nassim Kamar/Pr Robert Salvayre
Rapporteurs : Pr Pierre Marquet/Pr Christophe Mariat
Présentée et soutenue par Cindy Canivet
Le 7 mai 2009
Titre : Apport du suivi pharmacodynamique des immunosuppresseurs en transplantation
d'organes


J’adresse mes sincères remerciements aux membres du jury qui ont accepté de
juger ce travail :
Monsieur le Professeur Christophe Mariat, je suis très honorée de l’intérêt que
vous avez porté à mon travail et je vous remercie d’avoir accepté d’en être le
rapporteur.
Monsieur le Professeur Pierre Marquet, je tiens à vous remercier pour notre
collaboration et pour avoir accepté d’être le rapporteur de ce travail.
Je tiens également à remercier
Le Professeur Dominique Durand qui m’a permis de travailler en collaboration
avec son service et qui m’a fait confiance.
Le Professeur Lionel Rostaing : merci pour votre confiance et votre générosité
tout au long de ce travail, je suis heureuse de vous avoir compté parmi les
membres du jury.
Le Professeur Robert Salvayre et le Dr Anne Nègre-Salvayre : merci de m’avoir
accueilli au sein de votre équipe, de votre amabilité, et de votre soutien au cours
de ce travail.
Je tiens tout particulièrement à remercier le Docteur Nassim Kamar sans qui
tout ce travail n’aurait pu être réalisé : de ta disponibilité malgré ton emploi du
temps surchargé, de m’avoir supporté de longues heures dans ton bureau, merci
pour ton humour, et encore merci pour la confiance que tu m’as apportée et pour
tout ce que tu m’as appris
Je remercie
Le Dr Mogens Thomsen qui m’a accueillie initialement dans son équipe.
Le Dr Torsten Böhler : merci de m’avoir fait confiance, merci pour nos
conversations franco-germano-anglophones, pour ton réconfort et ta joie de
vivre.
Je remercie toute l’équipe 10,
Nathalie pour son humour, son réconfort, sa disponibilité et ses connaissances
scientifiques très précieuses, Cécile pour nos discussions variées, et ta
disponibilité, Françoise pour ton réconfort dans les moments de doutes.
Tous les étudiants avec qui j’ai partagé quelques repas et apéros, Marie qui a
bien souvent partagé avec moi les nocturnes et les we au labo, mais aussi
Raphael, Cécile, Aurélie, Carole, Elodie, et tous ceux qui sont passés au
laboratoire.
L’aide technique, Corinne pour avoir partagé les plaisirs du grand froid et qui ne
m’a jamais laissée sans PBS, Stéphanie pour son aide précieuse avec les
commandes, sa gentillesse et son soutien.

Un remerciement un peu spécial à mes deux DEA préférés Thomas pour nos fous
rires, ton humour décalé, et les bons petits plats de ta maman, Magalie pour
avoir partagé mon bureau de ministre, merci pour ta gentillesse et ta générosité
je suis sûre que tu seras une parfaite thésarde !
Merci à Sylvain, pour tout ce que l’on a partagé ensemble dans notre petite
équipe greffe, pour nos franches rigolades qui ont apporté à ces 3 années de
thèse de merveilleux souvenirs, merci pour ton aide et pour les WE que tu m’as
épargné, est-ce que tu élucideras le mystère de l’agrafeur fou ?
Merci à Eve et Danièle pour leur disponibilité et leur aide tout au long de ma
thèse, à Chantal pour sa patience et son organisation.
Merci à Jean-Claude Lepert pour son aide précieuse au cytomètre et pour
m’avoir permis de venir travailler soir et we.
Un grand grand merci à tous les infirmiers et infirmières qui ont fait que ce
travail soit possible aussi bien les filles de l’hématologie qui ont épargné à mes
petits bras de se faire trouer trop souvent et les filles de l’UTO qui m’ont
toujours accueillies avec gentillesse et ont su m’accepter malgré la surcharge de
travail que je leur ai apportées.
Un merci tout particulier à Nicole Chincholle et Noëlle, les infirmières de la
planification ainsi que Nicole et Patricia qui ont été d’une patience admirable,
merci de m’avoir épargné tous ces we!
Un grand merci à toute l’équipe 14 qui a dû supporter ma présence et mes
bavardages à la pause-café.
Merci à Nicole pour avoir partagé 1 an de PD avec moi, merci pour ton aide, nos
rigolades et ta gentillesse.
Merci à Bruno pour ton humour et ta sincérité, merci à Hervé pour m’avoir
accepté comme compagnon de bureau, ce fut des moments très agréables et
Boby Lapointe et Georges Brassens ne diront pas le contraire.
Merci à tous les autres membres de l’équipe, Virginie, Elodie, Nathalie, Stéphane,
Caroline, Fred, Thierry…
Merci à ma dévergondée, ton amitié dés mon arrivée, ton dynamisme, ta gaîté
m’ont fait aimer ma vie à Toulouse, rien n’aurait été pareil sans toi, j’espère que
tout cela continuera bien au-delà de la recherche.
Merci à Cosette, au caractère explosif, maintenant que tu n’habites plus chez les
Tavernier, j’espère que tu m’inviteras toujours pour ta blanquette ou ton poulet
basquaise ! Reste telle que tu es !

Merci à ma conductrice préférée, nos petites conversations au labo me manquent
déjà, heureusement que l’on va encore partager pleins de soirées animées ou pas
d’ailleurs car la grand-mère qui est en moi n’a pas encore cessé de te surprendre.
Merci à mon hôtesse de charme, pour ta franchise, tes dîners presque parfaits
et ton amitié, même si on ne se croisera plus dans les couloirs, j’espère bien
partager encore de nombreuses soirées et we avec toi.
Merci au saltimbanque, j’espère que l’on partagera encore de nombreuses
soirées.
Merci à mes parents et mes sœurs qui m’ont soutenu tout au long de ce travail,
merci à mes beaux-parents pour leur accueil chaleureux.
Merci à Sylvain, qui partage mon quotidien, mes joies, mes peines, mes difficultés
et qui accepte sans rien dire ma mauvaise humeur et me soutient dans les
moments de stress. Merci pour ta patience et tout l’amour que tu me portes,
j’espère que l’on partagera ce chemin encore longtemps…
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
1
/
161
100%