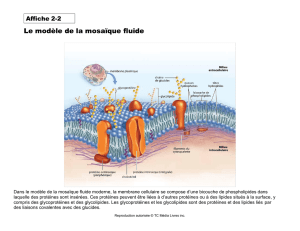
d-les cellules dendritiques

T
TH
HÈ
ÈS
SE
E
En vue de l'obtention du
D
DO
OC
CT
TO
OR
RA
AT
T
D
DE
E
L
L’
’U
UN
NI
IV
VE
ER
RS
SI
IT
TÉ
É
D
DE
E
T
TO
OU
UL
LO
OU
US
SE
E
Délivré par l'Université Toulouse III - Paul Sabatier
Discipline ou spécialité : Immunologie
JURY
Pr Salvatore Valitutti Président du Jury
Pr Jacques Urbain Rapporteur
Pr Thierry Velu Rapporteur
Dr Gervaise Loirand Examinatrice
Dr Anne Françoise Tilkin-Mariamé Directrice de Thèse
Pr Gilles Favre Co-directeur de Thèse
Ecole doctorale : Biologie-Santé-Biotechnologies
Unité de recherche : INSERM U563
Directeur(s) de Thèse : Dr Anne-Françoise Tilkin-Mariamé et le Pr Gilles Favre
Rapporteurs : Pr Jacques Urbain et Pr Thierry Velu
Présentée et soutenue par Guillaume SARRABAYROUSE
Le 14 décembre 2007
Titre : Rôle régulateur des protéines Rho dans la réponse Immune anti-mélanome.

Résumé
Les stratégies d’immunothérapie sont basées sur l’existence d’antigènes de tumeur (TAA)
contre lesquels l’hôte est capable d’induire une réponse immune spécifique. Cependant, les
cellules tumorales sont souvent peu immunogènes à cause de leur faible densité en
complexes CMH de classe I (CMH-I)/TAA ou à cause de leur incapacité à délivrer des
signaux de costimulation.
Au cours de ce travail, nous avons mis en évidence, que l’inhibition de la géranylgéranyl
transférase I par le GGTI-298 favorise l’induction d’une réponse immune anti-mélanome, en
induisant, sur les cellules tumorales murines B16F10 traitées, une expression des molécules
de costimulation (CD80 et CD86) et une surexpression des molécules du CMH-I, induite par
l’INF-γ. Ces modifications permettent de favoriser le rejet immun des mélanomes traités
injectés à des souris immunocompétentes et induisent chez ces souris l’activation de
lymphocytes T spécifiques. Comme différentes petites protéines G de la famille Rho peuvent
être géranylgéranylées, l’utilisation d’inhibiteurs spécifiques des protéines Rho (Rho A, Rho
B et Rho C) a montré leur(s) implication(s) dans ces régulations. Ainsi, le traitement in vitro
des cellules mélaniques murines par la combinaison d’INF-γ et d’inhibiteurs des protéines
Rho (C3 exoenzyme) induit une forte surexpression des molécules CMH-I et des molécules
de costimulation CD80 et CD86 à la membrane des cellules tumorales.
Par ailleurs, nous avons évalué l’intérêt thérapeutique de nos traitements pharmacologiques
associant l’IFN-γ et le GGTI-298. Nous avons dans un premier temps testé le potentiel
vaccinal de nos traitements en montrant que des souris injectées avec des cellules B16F10
préalablement traitées et iradiées étaient partiellement protégées contre le développement
de tumeurs sauvages. Dans un deuxième temps, nous avons montré que ces traitements
induisent sur des tumeurs humaines une surexpression des molécules HLA de classe I et
des molécules de costimulation ayant des activités activatrices (CD86) ou inhibitrices (PD-
L1) et provenant des familles B7 ou TNF/TNFR. De plus, nous avons mis en évidence la
capacité stimulatrice de ces mélanomes humains prétraités à induire in vitro l’activation de
lymphocytes T CD8 cytotoxiques spécifiques des mélanomes et sécrétant de l’IFN−γ, à partir
de donneurs sains HLA-compatibles.
Au cours de cette thèse nous avons également mis en évidence le rôle régulateur des
protéines Rho dans l’expression de deux molécules de la famille des TNF : « FasL et
CD70 ». La molécule FasL est exprimée sur de nombreuses lignées tumorales et favorise le
mécanisme de contre-attaque tumorale en induisant l’apoptose de LT ou LB Fas positifs par
la voie d’apoptose induite par la fixation du ligand Fas-L sur son récepteur Fas. Nous avons
montré que la protéine RhoA et son effecteur ROCKp160 régulent de façon négative
l’expression de Fas-L à la membrane des cellules de mélanome murin B16F10. Ces
protéines limitent ainsi in vitro le mécanisme de contre-attaque tumorale. La molécule CD70
est exprimée sur diverses lignées tumorales mais son rôle fait encore l’objet de
controverses. Nous avons montré pour la première fois qu’une lignée de mélanome
métastatique humaine exprime de façon constitutive la molécule CD70 et que cette
expression, régulée positivement par la protéine RhoA, inhibe in vitro la prolifération de
lymphocytes allogéniques.
L’ensemble de ce travail de thèse a permis de montrer que les protéines Rho régulent
l’expression de nombreuses molécules impliquées dans la mise en place de réponses
immunes anti-mélanome. Ces résultats permettent d’envisager le développement de
protocoles d’immunothérapie clinique utilisant des cellules de mélanomes HLA-compatibles
prétraitées in vitro par l’association IFNγ plus GGTI-298 comme cellules « thérapeutiques »
ou vaccinales.

Summary
Melanoma is a highly lethal cutaneous tumor, killing affected patients through multiple, poorly
immunogenic metastases. Suboptimal activation of T lymphocytes by melanoma cells is
often due to the defective expression of class I major histocompatibility antigens (MHC-I) and
costimulatory molecules, or to the capacity of FasL molecules expressed on melanoma cells
to induce lymphocyte apoptosis.
In this work, I demonstrated that statins, inhibitors of HMGCoA reductase, enhance mIFN-γ
induced expression of MHC class I antigens on murine B16F10 melanoma. GGTI-
298, a geranylgeranyl transferase I inhibitor, mimics this effect of statins that is related to
peptide transporter protein TAP1 up-regulation. Simultaneously, GGTI-298 induces the
expression of CD80 and CD86 costimulatory molecules. C3 exoenzyme, which selectively
inactivates Rho proteins, phenocopies the effects of GGTI-298, indicating a role for Rho
proteins in these events. Furthermore, the treatment of B16F10 cells with GGTI-298 or C3
exoenzyme associated with mIFN-gamma induces in vivo tumor growth slowing down in
immunocompetent but not in nu/nu syngeneic mice. Both in vivo injections and in vitro
restimulation of splenocytes with GGTI-298- and mIFN-gamma-treated B16F10 cells induces
an enhancement of specific CD8 T lymphocytes labeled by TRP-2/H-2K(b) tetramers. Using
a human melanoma model (LB1319-MEL), we demonstrated that in vitro treatment with
hIFN-γ and GGTI-298 led to the up regulation of MHC-I and a costimulatory molecule CD86
and down regulation of an inhibitory molecule PD-1L. Co-culture experiments with peripheral
blood mononuclear cells (PBMC) revealed that modifications induced by hIFN-! and GGTI-
298 on the selected melanoma cells, enables the stimulation of lymphocytes from HLA
compatible healthy donors. Indeed, as compared with untreated melanoma cells,
pretreatment with hIFN-γ and GGTI-298 together rendered the melanoma cells more efficient
at inducing the: i) activation of CD8 T lymphocytes (CD8+/CD69+); ii) proliferation of tumor-
specific CD8 T cells (MelanA-MART1/TCR+); iii) secretion of hIFN-γ; and iv) anti-melanoma
specific cytotoxic cells. In this study, it is shown that vaccination with mIFN-γ and GGTI-298
pretreated B16F10 cells induces a protection against untreated tumor growth and pulmonary
metastases implantation.
Furthermore the capacity of FasL molecules expressed on melanoma cells to induce
lymphocyte apoptosis contributes to either antitumor immune response or escape depending
on their expression level. Little is known, however, about the mechanisms regulating FasL
protein expression. Using the murine B16F10 melanoma model weakly positive for FasL, we
demonstrated that in vitro treatment with statins enhances membrane FasL expression. C3
exotoxin and the geranylgeranyl transferase I inhibitor GGTI-298, mimic this effect. Inhibition
of RhoA expression by small interfering RNA (siRNA) increased membrane FasL expression,
whereas overexpression of constitutively active RhoA following transfection of RhoAV14
plasmid decreased it. Moreover, the inhibition of a RhoA downstream effector p160ROCK
also induced this FasL overexpression. We conclude that the RhoA/ROCK pathway
negatively regulates membrane FasL expression in these melanoma cells. Furthermore, we
have shown that B16F10 cells, through the RhoA/ROCK pathway, promote in vitro apoptosis
of Fas-sensitive A20 lymphoma cells.
All together, these data indicate that protein geranylgeranylation as well as Rho protein are
critical for regulating on melanomas the expresion of molecules involved in anti-melanoma
immune response. These results, indicate that treatment of melanoma cell lines with
pharmacological molecules targeting Rho proteins, be a novel approach to produce tumor
cells suitable for vaccination and for stimulation of anti-melanoma effector cells.

SOMMAIRE
Données Bibliographiques………………………………………………..p1
Partie I : La réponse immune anti-tumorale et ses limites
I-La réponse immune innée anti-tumorale………………………………...p3
A-Les cellules Natural Killer : NK
B-Les lymphocytes Natural Killer T : NKT
C-Les lymphocytes T gamma-delta : LTγδ
D-Les Cellules Dendritiques : Les CD
1) La capture de l’antigène
2) La dégradation et l’apprêtement des antigènes
2.1) Voie de présentation par les molécules du CMH-I
2.2) Voie de présentation par les molécules du CMH-II
2.3) Voie alternative, présentation croisée
3) Migration et maturation des CD
4) Les cytokines et chémokines
E-Les Cellules dendritiques myéloïdes (CDm) et plasmacytoïdes (CDp)
F-Les IKDC le chaînon manquant
II-La réponse immune adaptative anti-tumorale………………………...p25
A-Les Antigènes de tumeurs
1) Les antigènes d’origine embryonnaire
2) Les antigènes de différenciation
3) Les antigènes surexprimés
4) Les antigènes spécifiques des tumeurs
B-L’activation de la réponse immune adaptative
1) La signalisation par le TCR : le premier signal d’activation lymphocytaire
2) Le signal de costimulation
2-1) Les molécules de costimulation de la famille des immunoglobulines:
2-2) Les molécules de costimulation de la famille des TNF (Tumor Necrosis Factor)
2-3) Les ligands activateurs des cellules NK
3) La synapse immunologique
C-Acquisition des fonctions effectrices des LT et rôles dans la réponse
immune anti-tumorale.
1) Les LTCD8 cytotoxiques
2) Les LTCD4 auxiliaires
D-La mémoire immunitaire associée aux LT
E-Les Lymphocytes B
III Mécanismes d’échappement des tumeurs à
l’immunosurveillance……………………………………………………………...p44
A-Mécanismes d’échappement tumoral lié à des modifications membranaires
des cellules tumorales
6) Perte ou diminution de l’expression des molécules du CMH-I
2) La contre attaque tumorale associée à l’expression de protéines de la famille du TNF
3) Rôle de deux molécules inhibitrices de la famille des immunoglobulines dans l’échappement
tumoral : les protéines PD-L1 et PD-L2
4) Rôle des molécules de CMH non classique dans l’échappement tumoral : les protéines HLA-
G et HLA-E
B-Mécanismes d’échappement tumoral associés aux effecteurs immuns
6) Les LT régulateurs : les Treg
2) Les Macrophages Associés aux Tumeurs : Les TAM
3) Les cellules dendritiques : Les CD
3.1) Les cellules dendritiques immatures (Cdi)
3-2) Les cellules dendritiques plasmacytoïdes : (CDp)

4) Les cellules suppresseurs myéloïdes : Les MSC
C-Les Mécanismes d’échappement tumoral associés auxmolécules du
microenvironnement
1)L’ indoleamine 2,3-dioxygenase : IDO
2) Le facteur de croissance transformant béta : TGFβ
3) Le VEGF (Vascular Endothelial Growth Factor):
4) Les Chémokines
5) La cyclooxygénase 2 : COX2
6) L’arginase et la nitric-oxide synthase : ARG et NOS
Partie II : Les protéines Rho de nouvelles cibles
thérapeutiques dans l’immunothérapie du mélanome
I Le mélanome cutané…………………………………………………………...p60
A-Les divers types de mélanomes
B-Histologie et développement des mélanomes
C-Altérations génétiques impliquées dans l’apparition des mélanomes
D) La voie de signalisation Ras / Raf / MEK / Erk
2) Le facteur de transcription MITF
3) La voie de la PI3K
4) Mutations des voies de survie et d’apoptose
5)Altérations des gènes suppresseurs de tumeur
D-Traitements ciblés des mélanomes
II Immunothérapies du mélanome……………………………………………p72
A-L’Interféron alpha (IFN-α) et l’Interleukine 2 (IL-2)
B-La chimio-immunothérapie
C-Les immunoadjuvants
D-La vaccination par cellules tumorales
E-La vaccination par protéines de choc thermiques
F-La vaccination par peptides ou protéines de tumeurs
G-La Vaccination par cellules dendritiques
H-Vaccination par plasmides ou vecteurs viraux
I-L’immunothérapie adoptive
J-Electrochimiothérapie et électroimmunothérapie
III Les statines………………………………………………………………………..p83
A-Origine et mode d’action des statines
B-Statines et cancer
1) Lignées de cancers colorectaux
2) Lignées de cancers du sein
3) Lignées de mélanomes
C-Rôles immunomodulateurs des statines
IV Les petites protéines G de la famille Rho……………………………………...p91
A-Propriétés générales des protéines Rho
B-Fonctions des protéines Rho
1) GTPases Rho et cytosquelette
1-a) GTPases Rho et cytosquelette d’actine
1-b) GTPases Rho et microtubules
2) GTPases Rho et trafic cellulaire
3) GTPases Rho et expression génique
4) GTPases Rho et progression du cycle cellulaire
5) GTPases Rho et apoptose
C-Propriétés immunomodulatrices des protéines Rho
1) GTPases Rho et sélection thymique
2) GTPases Rho et activation lymphocytaire
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
1
/
187
100%