MET, une nouvelle cible thérapeutique dans le cancer bronchique

Correspondances en Onco-Théranostic - Vol. II - n° 2 - avril-mai-juin 2013
93
Mise au point
MET, une nouvelle cible thérapeutique
dans le cancer bronchique
MET, a novel therapeutic target in lung cancer
Anne-Marie Ruppert1, 2, Armelle Lavolé1, 2, Martine Antoine3, Marie Wislez1, 2, Jacques Cadranel1, 2
1. Service de
pneumologie, hôpital
Tenon, AP-HP, Paris.
2. Équipe de recherche2,
GRC-UPMC 04
Theranoscan, université
Pierre-et-Marie-Curie,
université Paris-VI.
3. Service d’anatomie
pathologique, hôpital
Tenon, AP-HP, Paris.
RÉSUMÉ
Summary
»
MET est un récepteur membranaire à activité tyrosine kinase
dont le ligand est le facteur de croissance hépatocytaire (HGF),
également appelé “scatter factor”. L’activation de la voie HGF/MET
induit dans les cellules des propriétés mitogènes, motogènes
et proangiogéniques essentielles au cours du développement
embryonnaire, mais également impliquées dans le processus
cancéreux. Son activation au cours du cancer bronchique serait
associée à un mauvais pronostic. Différentes anomalies de la voie
ont été décrites : surexpression du ligand HGF, surexpression
du récepteur, amplification génomique ou mutations de MET.
En fonction du type d’altération, dans les tumeurs où MET est
amplifié ou muté, MET serait un oncogène. Dans les autres cancers
bronchiques, MET serait impliqué dans la progression tumorale
par invasion tissulaire et formation de métastases. Par ailleurs,
l’amplification de MET est un mécanisme de résistance secondaire
connu, retrouvé chez environ 20 % des patients présentant un
cancer bronchique muté pour EGFR et traité par inhibiteur de
tyrosine kinases d’EGFR. Différentes stratégies d’inhibition de MET
sont en cours de développement dans le cancer bronchique, en
particulier chez les patients en progression après une réponse
initiale sous inhibiteur d’EGFR.
Mots-clés : MET – HGF – Cancer bronchique – Pronostic – Biomarqueur –
Récepteur de tyrosine kinases – Inhibiteur de tyrosine kinases.
MET is a cell membrane tyrosine kinase receptor for its ligand,
the hepatocyte growth factor (HGF), also called scatter factor
(SF). MET presents mitogenic, motogenic and pro-angiogenic
signals which are essential during embryonic development
and during cancer progression. Activation of the HGF/MET
pathway seems to be associated with a worse prognosis in
lung cancer. Several molecular anomalies of the pathway
are reported in lung cancer: ligand overexpression, receptor
overexpression, genomic amplification or MET mutation.
In MET amplified or mutated lung cancer, MET seems to
be an oncogene as the tumor appears to be MET addicted.
In other lung cancers, MET may be implicated in tumor
progression by tissue invasion and formation of metastases.
MET amplification is also a known mechanism implicated
in 20% of secondary resistance to EGFR inhibitors in patients
presenting EGFR mutated lung cancer. Different strategies of
MET inhibition in lung cancer are being studied, particularly
in EGFR mutated lung cancer.
Keywords: MET – HGF – Lung cancer – Prognosis – Biomarker
– Tyrosine kinase receptor – Tyrosine kinase inhibitor.
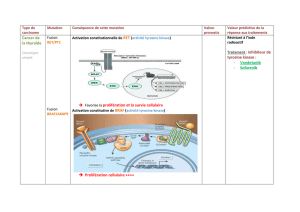
Biologie de MET
Le gène du récepteur MET est situé sur le bras long
du chromosome 7 (7q31) [1, 2]. Le récepteur MET fait
partie de la famille des récepteurs de tyrosine kinases
et comprend une sous-unité α extracellulaire et une
sous-unité β composée de 3 parties différentes : extra-
cellulaire, transmembranaire et intracellulaire (figure 1,
p. 94). L’HGF est le ligand exclusif de MET. Il est sécrété
majoritairement par les cellules mésenchymateuses
du stroma et les polynucléaires neutrophiles (figure 2,
p. 94).
Le récepteur est inactif sous sa forme de monomère.
Après s’être lié à son ligand, le HGF, MET forme soit des
homodimères, soit des hétérodimères avec d’autres
récepteurs de tyrosine kinases pouvant activer les voies
de signalisation, des Ras/MAP kinases, de PI3K-AKT/
mTOR et de JAK (JAnus Kinase)/STAT (Signal Transducer
and Activator of Transcription) [1].
La voie MET/HGF joue un rôle clé au cours de l’embryo-
genèse (1, 2) sur la migration, la différenciation, la pro-
lifération et la survie cellulaires ; elle est associée au
phénomène de “transition épithéliomésenchymateuse”
des cellules. Cette croissance invasive est quiescente

Correspondances en Onco-Théranostic - Vol. II - n° 2 - avril-mai-juin 2013
94
Mise au point
chez l’adulte. Elle peut être réactivée en cas de lésions
tissulaires, lorsque les cellules résiduelles doivent pro-
liférer et migrer pour restituer l’intégrité du tissu. Ainsi,
le taux plasmatique d’HGF augmente après une lésion
hépatique, rénale ou pulmonaire.
MET/HGF et cancer bronchique
La voie MET/HGF est impliquée dans l’oncogenèse de
différentes tumeurs solides. MET peut jouer un rôle
clé dans certains cancers, comme le cancer papillaire
du rein. Des mutations ponctuelles dans le domaine
tyrosine kinase de MET y sont retrouvées, que ce soit
dans les formes sporadiques ou héréditaires (4). Ces
mutations aboutissent à une activation constitutive du
récepteur et favorisent le développement de cancers
du rein souvent multiples et bilatéraux. Ces cancers
mutés présentent une addiction oncogénique vis-à-vis
de MET (4). Les mutations ponctuelles de MET sont
identifiées dans le cancer bronchique au niveau de
séquences codant pour le domaine juxtamembra-
naire, région régulatrice du récepteur tyrosine kinase,
et au niveau du domaine Sema, site de fixation de
l’HGF (5-7). Ces mutations augmentent in vitro la pro-
lifération et la motilité cellulaires (7). Leur rôle in vivo
reste à déterminer.
Le nombre de copies de MET peut être évalué par FISH
et par PCR quantitative. Un nombre élevé de copies du
gène MET (≥ 5 copies/cellule) est retrouvé dans environ
11 % des cancers bronchiques (8, 9). L’augmentation
du nombre de copies de MET est plus fréquente dans
les adénocarcinomes peu différenciés et associée à un
pronostic défavorable (8, 9).
La surexpression protéique de MET est associée à un
pronostic défavorable dans de multiples cancers. MET
est impliqué dans la progression tumorale au cours
des cancers du côlon, du sein ou de l’ovaire (1). Le site
www.vai.org/met rapporte les principales références des
publications concernant MET dans les différents cancers.
La surexpression de MET est fréquente dans le cancer
bronchique. Elle est retrouvée plus souvent dans les
adénocarcinomes (27 à 67 %) que les carcinomes épider-
moïdes (0,7 à 57 %) et les cancers bronchiques à petites
cellules (25 %) [7, 9]. La surexpression de MET est associée
à celle de son récepteur activé, la forme phosphorylée
active de MET (p-MET) [7, 9]. La surexpression de p-MET
est surtout observée dans les cancers bronchiques non
à petites cellules (CBNPC) exprimant intensément MET
et l’HGF (10). La surexpression de MET et p-MET serait
impliquée dans le développement des adénocarcinomes
pulmonaires de type papillaire (10, 11). L’effet pronostique
Figure 1. Structure de MET
(3)
.
α β
SS
SEMA
Séquence liée à MET
Domaine kinase
Structure
immunoglobulin-like
Domaine extracellulaire
Domaine juxtamembranaire
Domaine intracellulaire
Ser 985
Tyr 1003
Tyr 1234
Tyr 1235
Tyr 1349
Tyr 1356 Site de fixation
MET
Figure 2. L’HGF.
N K1 K2 K3 K4 Domaine sérine protéase
RVV
9-9
HGF

Correspondances en Onco-Théranostic - Vol. II - n° 2 - avril-mai-juin 2013
95
MET, une nouvelle cible thérapeutique dans le cancer bronchique
péjoratif de la surexpression de MET reste discuté. Seules
2 études ont montré que la surexpression tumorale de
MET est un facteur pronostique défavorable (12, 13).
L’effet pronostique défavorable de la surexpression de
MET est confirmé par les données obtenues à partir de
xénogreffes de lignées cellulaires tumorales surexprimant
MET et/ou l’HGF. En cas d’expression à la fois de MET
et de son ligand, la tumorigénicité est accrue, avec un
risque élevé de développement de métastases (14, 15).
Par ailleurs, un taux élevé d’HGF tumoral est un facteur
pronostique défavorable indépendant au cours des adé-
nocarcinomes (16, 17). L’HGF est essentiellement sécrété
par les cellules du stroma (sécrétion paracrine), mais
peut être sécrété directement par la cellule tumorale
(sécrétion autocrine).
MET comme mécanisme de résistance
aux inhibiteurs d’EGFR
MET est impliqué dans les mécanismes de résistance
acquise des cancers bronchiques mutés pour EGFR
progressant après une réponse initiale aux inhibiteurs
de la fonction tyrosine kinase de l’EGFR (ITK-EGFR). Une
amplification de MET est retrouvée dans environ 20 %
des tumeurs mutées pour EGFR (18). Il y a dans ce cas
une hétérodimérisation de MET et de HER3 permettant
de déjouer les ITK-EGFR, HER3 n’ayant pas de domaine
tyrosine kinase. De façon intéressante, l’inhibition de
MET rétablit dans les modèles animaux la sensibilité à
l’erlotinib (19).
MET comme cible thérapeutique
Pour MET, 2 schémas de traitement doivent être dis-
tingués.
✓
D’une part, l’amplification et les mutations de MET
sont des altérations génomiques rares, qui pourraient
constituer une cible thérapeutique d’addiction oncogé-
nique (20). Les lignées tumorales amplifiées pour MET
sont dépendantes de MET, et l’inhibition du récepteur
aboutit à un arrêt de la prolifération et à une mort par
apoptose (20). Peu de données sont disponibles in vivo.
Un cas clinique rapporte l’efficacité d’un inhibiteur de
MET, le crizotinib, sur un cancer bronchique amplifié
pour MET (21). MET est aussi un mécanisme de résistance
acquise des cancers bronchiques mutés pour EGFR pro-
gressant après une réponse initiale aux inhibiteurs de la
fonction tyrosine kinase d’EGFR. L’association d’un inhi-
biteur de MET avec la poursuite de l’ITK-EGFR constitue
une approche thérapeutique possible chez ces malades.
✓
D’autre part, les inhibiteurs de MET ont démontré
leur efficacité sur de nombreuses lignées cellulaires,
alors qu’aucune altération génomique, mutation ou
amplification, n’a pu être mise en évidence. Dans une
étude expérimentale, MET surexprimé dans les lignées
cellulaires puis dans des xénogreffes de cellules tumo-
rales était inhibé par un brin d’ARN interférant (siRNA)
[22]. Cela aboutissait à l’arrêt de la croissance invasive
et de la prolifération in vitro, puis à la régression des
métastases in vivo.
Les différentes stratégies d’inhibition
de la voie MET/HGF(23)
Inhibition du ligand HGF par un anticorps
anti-HGF humanisé (AMG 102)
Cet anticorps a été évalué au cours de 2 études de
phase I/II en association avec l’erlotinib, dans le can-
cer bronchique non à petites cellules prétraité, et en
association avec le cisplatine et l’étoposide, dans le
cancer bronchique à petites cellules (NCT00791154
et NCT01233687).
Inhibition du récepteur MET par un anticorps
monoclonal humanisé anti-MET (onartuzumab ;
Genentech)
Dans une étude de phase II (NCT00854308) compor-
tant 128 patients présentant un cancer bronchique
non à petites cellules de stade IIIb/IV, un traitement
par erlotinib 150 mg associé à cet anticorps, l’onar-
tuzumab (15 mg/kg/semaine), ou à un placebo a
été administré en deuxième ou troisième ligne (24).
L’expression de MET a été recherchée systématique-
ment dans les prélèvements tumoraux par immu-
nohistochimie ; elle était observée dans 56,5 % des
tumeurs des patients du groupe onartuzumab et
50,8 % des tumeurs des patients du groupe placebo.
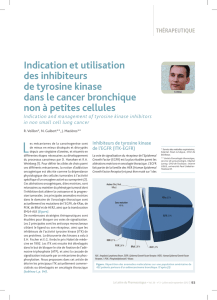
La survie sans progression dans la cohorte en inten-
tion de traiter était comparable dans les 2 groupes. En
revanche, l’analyse de la population présentant une
hyperexpression de MET montre un effet bénéfique
sur la survie sans progression (7,7 versus 7,4 mois ;
HR = 0,55 ; IC95 : 0,26-1,16) et la survie globale (12,4
versus 8,4 mois ; HR = 0,56 ; IC95 : 0,31-1,02). Cette
étude montre, dans le bras placebo, l’influence pro-
nostique défavorable de l’hyperexpression de MET.
Une étude de phase III (NCT01456325) est actuel-
lement en cours en deuxième ligne dans le CBNPC
avec hyperexpression de MET : l’association erlotinib
+ onartuzumab est comparée à l’association erlotinib
+ placebo.

Correspondances en Onco-Théranostic - Vol. II - n° 2 - avril-mai-juin 2013
96
Mise au point
Les auteurs déclarent
nepas avoir de liens
d’intérêts.
Inhibition de MET par un inhibiteur de tyrosine
kinases sélectif
Différentes molécules (ARQ197, JNJ-38877605,
PF-04217903) sont en cours d’évaluation. L’ARQ197 a
montré un profil de tolérance et d’efficacité acceptable
en phase I. Son association in vitro avec l’erlotinib montre
une efficacité supérieure à celle de chacune des molécules
prises séparément. Les résultats d’une étude de phase II
d’ARQ197 ont été présentés au congrès de l’European
Society for Medical Oncology (ESMO) en 2010 (25). Dans
cet essai incluant des CBNPC de stade avancé en deu-
xième ligne ou plus, 167 patients ont été randomisés en
un groupe erlotinib (150 mg/j) + ARQ197 (360 mg × 2/j) et
un groupe erlotinib + placebo. La survie sans progression,
objectif principal, était significativement allongée dans le
bras ARQ197 : 3,7 versus 2,2 mois (HR = 0,68 ; IC95 : 0,47-
0,98 ; p < 0,05). L’amélioration de la survie sans progression
semble meilleure pour l’histologie non épidermoïde et
pour les tumeurs à EGFR sauvage, avec amplification de
MET ou avec mutation de Ras. Les résultats d’une étude de
phase III (NCT01244191) évaluant l’ARQ197 en association
avec l’erlotinib en deuxième ou troisième ligne dans le
CBNPC non épidermoïde sont attendus prochainement.
Une étude de phase II (NCT01395758) est actuellement
en cours dans les CBNPC mutés pour Ras.
Inhibition de MET par crizotinib,
un inhibiteur multikinase (PF-02341066)
ciblant à la fois ALK et MET
Le crizotinib a actuellement une AMM en deuxième
ligne dans les cancers bronchiques ayant une translo-
cation d’ALK. Les données in vitro du PF-02341066 sur
des lignées amplifiées pour MET sont intéressantes : il
inhibe de la prolifération et la migration cellulaires. Peu de
données sont disponibles in vivo. Plusieurs cas cliniques
rapportent des réponses spectaculaires dans des cancers
amplifiés pour MET, dont 1 cas de CBNPC (21).
Inhibition de MET par des inhibiteurs
de tyrosine kinases à large spectre
(BMS907351 ; GSK1363089)
Le BMS907351 est un inhibiteur de MET, VEGFR2 et
RET qui a été évalué au cours d’une étude de phase
II dans différents cancers solides, dont le CBNPC.
Même si le nombre de patients atteints de CBNPC est
faible, les résultats sur les métastases viscérales sont
intéressants : certaines lésions régressent. Une étude
de phase I/II (NCT00596648) est en cours. Elle évalue
le BMS907351 en association avec l’erlotinib dans le
CBNPC. Le GSK1363089 est un inhibiteur ciblant MET,
VEGFR1-3, RET, KIT et FIT-3 qui a montré son efficacité
dans des modèles précliniques de cancer bronchique.
Une étude de phase I-II est en cours (NCT01068587).
Perspectives
Des progrès indéniables ont été réalisés au cours de cette
décennie concernant la compréhension de l’implication
de l’oncogène MET au cours du cancer du poumon. Les
inhibiteurs de MET ont été développés dans de multiples
situations, avec ou sans ITK-EGFR, dans des CBNPC mutés
ou non pour EGFR. Les résultats préliminaires encoura-
geants des essais des inhibiteurs de MET nous confortent
dans la nécessité de poursuivre le développement des
thérapeutiques ciblées “à la carte”. ■
1. Trusolino L, Bertotti A, Comoglio PM. MET signalling: principles and
functions in development, organ regeneration and cancer. Nat Rev
Mol Cell Biol 2010;11(12):834-48.
2. Ruppert AM, Beau-Faller M, Belmont L et al. Un regard simple sur la
biologie du cancer bronchique : MET. Rev Mal Respir 2011;30(4):1201-2.
3.
Benvenuti S, Comoglio PM. The MET receptor tyrosine kinase in
invasion and metastasis. J Cell Physiol 2007;213(2):316-25.
4. Schmidt L, Duh FM, Chen F et al. Germline and somatic mutations
in the tyrosine kinase domain of the MET proto-oncogene in papillary
renal carcinomas. Nat Genet 1997;16(1):68-73.
5. Ma PC, Kijima T, Maulik G et al. c-MET mutational analysis in
small-cell lung cancer: novel juxtamembrane domain mutations
regulating cytoskeletal functions. Cancer Res 2003;63(19):6272-81.
6. Kong-Beltran M, Seshagiri S, Zha J et al. Somatic mutations
lead to an oncogenic deletion of met in lung cancer. Cancer Res
2006;66(1):283-9.
7. Ma PC, Jagadeeswaran R, Jagadeesh S et al. Functional expression
and mutations of c-Met and its therapeutic inhibition with SU11274
and small interfering RNA in non-small-cell lung cancer. Cancer Res
2005;65(4):1479-88.
8.
Cappuzzo F, Marchetti A, Skokan M et al. Increased MET gene
copy number negatively affects survival of surgically resected non-
small-cell lung cancer patients. J Clin Oncol 2009;27(10):1667-74.
9.
Tsuta K, Kozu Y, Mimae T et al. c-MET/phospho-MET protein expres-
sion and MET gene copy number in non-small cell lung carcinomas.
J Thorac Oncol 2012;7(2):331-9.
10.
Nakamura Y, Niki T, Goto A et al. c-Met activation in lung ade-
nocarcinoma tissues: an immunohistochemical analysis. Cancer
Sci 2007;98(7):1006-13.
11. Tsao MS, Liu N, Chen JR et al. Differential expression of Met/
hepatocyte growth factor receptor in subtypes of non-small cell
lung cancers. Lung Cancer 1998;20(1):1-16.
12.
Masuya D, Huang C, Liu D et al. The tumour-stromal interaction
between intratumoral c-Met and stromal hepatocyte growth factor
associated with tumour growth and prognosis in non-small-cell
lung cancer patients. Br J Cancer 2004;90(8):1555-62.
13. Takanami I, Tanana F, Hashizume T et al. Hepatocyte growth
factor and c-Met/hepatocyte growth factor receptor in pulmonary
adenocarcinomas: an evaluation of their expression as prognostic
markers. Oncology 1996;53(5):392-7.
14. Navab R, Liu J, Seiden-Long I et al. Co-overexpression of Met
and hepatocyte growth factor promotes systemic metasta-
sis in NCI-H460 non-small cell lung carcinoma cells. Neoplasia
2009;11(12):1292-300.
15. Yi S, Tsao MS. Activation of hepatocyte growth factor-met
autocrine loop enhances tumorigenicity in a human lung ade-
nocarcinoma cell line. Neoplasia 2000;2(3):226-34.
16.
Siegfried JM, Luketich JD, Stabile LP, Christie N, Land SR. Elevated
hepatocyte growth factor level correlates with poor outcome in
early-stage and late-stage adenocarcinoma of the lung. Chest
2004;125(5 Suppl.):116S-9S.
17. Wislez M, Rabbe N, Marchal J et al. Hepatocyte growth factor
production by neutrophils infiltrating bronchioloalveolar subtype
pulmonary adenocarcinoma: role in tumor progression and death.
Cancer Res 2003;63(6):1405-12.
18. Engelman JA, Zejnullahu K, Mitsudomi T et al. MET amplification
leads to gefitinib resistance in lung cancer by activating ERBB3
signaling. Science 2007;316(5827):1039-43.
19.
Tang Z, Du R, Jiang S et al. Dual MET-EGFR combinatorial inhibi-
tion against T790M-EGFR-mediated erlotinib-resistant lung cancer.
Br J Cancer 2008;99(6):911-22.
20.
Lutterbach B, Zeng Q, Davis LJ et al. Lung cancer cell lines harbor-
ing MET gene amplification are dependent on Met for growth and
survival. Cancer Res 2007;67(5):2081-8.
21. Ou SHI, Kwak EL, Siwak-Tapp C et al. Activity of crizotinib
(PF02341066), a dual mesenchymal-epithelial transition (MET)
and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small-
cell lung cancer patient with de novo MET amplification. J Thorac
Oncol 2011;6(5):942-6.
22. Corso S, Migliore C, Ghiso E, De Rosa G, Comoglio PM, Giordano
S. Silencing the MET oncogene leads to regression of experimental
tumors and metastases. Oncogene 2008;27(5):684-93.
23. Feng Y, Thiagarajan PS, Ma PC. MET signaling: novel targeted
inhibition and its clinical development in lung cancer. J Thorac Oncol
2012;7(2):459-67.
24.
Spigel D. Randomized multicenter double-blind placebo-control-
led phase 2 study evaluating MetMAb, an antibody to Met receptor, in
combination with erlotinib, in patients with advanced non-small-cell
lung cancer. Program and abstracts of the 35th European Society
of Medical Oncology Congress, 8-12 octobre 2010, Milan, Italie.
Abstract LBA15.
25. Sequist L, Akerley W, Brugger W. Final results from ARQ 197-209:
a global randomized, placebo-controlled phase 2 clinical trial of
erlotinib plus ARQ 197 versus erlotinib plus placebo in previously
treated EGFR-inhibitor naïve patients with advanced non-small-cell
lung cancer (NSCLC). Program and abstracts of the 35th European
Society of Medical Oncology Congress, 8-12 octobre 2010, Milan,
Italie. Abstract 3630.
Références
1
/
4
100%