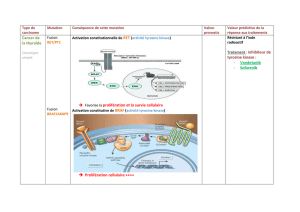
Lire l`article complet

THÉRAPEUTIQUE
R. Veillon
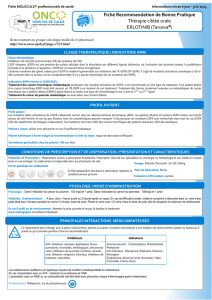
Figure. Répartition des anomalies moléculaires sur une population américaine de
422 patients porteurs d’un adénocarcinome bronchique. D’après (3).
ALK : Anaplastic Lymphoma Kinase ; EGFR : Epidermal Growth Factor Receptor ; HER2 : Human Epidermal Growth Factor
Receptor 2 ; Pi3K : Phosphatidylinositol 3-Kinase.
Autres (Met, Mek,
NRas)
Sans mutation
40 %
HER 0,7 %
Pi3K 2,6 % BRaf 2,8 % ALK 3,3 %
EGFR 23 %
KRas 25 %
La Lettre du Pharmacologue • Vol. 26 - n° 3 - juillet-août-septembre 2012 | 93
Indication et utilisation
des inhibiteurs
de tyrosine kinase
dans le cancer bronchique
non à petites cellules
Indication and management of tyrosine kinase inhibitors
in non small cell lung cancer
R. Veillon*, N. Guibert**, J. Mazières**
* Service des maladies respiratoires,
hôpital Haut-Lévêque, CHU de
Bordeaux.
** Unité d’oncologie thoracique,
service de pneumologie, hôpital
Larrey, CHU de Toulouse ; Inserm
U563, université Paul-Sabatier-
Toulouse III.
L
es mécanismes de la cancérogenèse sont
de mieux en mieux disséqués et décryptés
depuis une vingtaine d’années, et résumés en
différentes étapes nécessaires au développement
du processus cancéreux par D. Hanahan et R.A.
Weinberg (1). Pour définir les cibles de choix parmi
ces différents mécanismes, la notion d’addiction
oncogénique est décrite comme la dépendance
physiologique des cellules tumorales à l’activité
spécifique d’un oncogène activé ou surexprimé (2).
Ces altérations oncogéniques, dites motrices, sont
nécessaires au maintien du phénotype tumoral dont
l’inhibition doit altérer la croissance et la progres-
sion tumorales. Les principales anomalies motrices
dans le domaine de l’oncologie thoracique sont
actuellement les mutations de l’EGFR, de KRas, de
PI3K, de BRaf et de HER2, ainsi que la translocation
EML4-ALK (figure).
De nombreuses stratégies thérapeutiques sont
étudiées pour bloquer ces voies de signalisation.
Les 2 principales sont les anticorps monoclonaux
ciblant le ligand ou son récepteur, ainsi que les
inhibiteurs de l’activité tyrosine kinase (ITK) de
ces protéines. La découverte des kinases a valu à
E.H. Fischer et E.G. Krebs le prix Nobel de méde-
cine en 1992. Les ITK ont ensuite été développés
dans le but de bloquer le site de fixation de l’adé-
nosine triphosphate (ATP), et ainsi la cascade de
signalisation instaurée par ce mécanisme de phos-
phorylation. Nous proposons dans cet article de
décrire les principaux ITK actuellement commer-
cialisés ou développés en oncologie thoracique
(tableau I, p. 94).
Inhibiteurs de tyrosine kinase
de l’EGFR (ITK-EGFR)
La voie de signalisation du récepteur de l’Epidermal
Growth Factor (EGFR) est la plus étudiée parmi les
altérations motrices en oncologie thoracique. L’EGFR
fait partie de la famille des HER (Human Epidermal
Growth Factor Receptor) et peut être muté sur 1 des

94 | La Lettre du Pharmacologue • Vol. 26 - n° 3 - juillet-août-septembre 2012
Résumé
Les inhibiteurs de tyrosine kinase (ITK) sont entrés depuis 2005 dans la pratique courante en oncologie
thoracique, obligeant les oncologues à s’adapter à leurs propriétés pharmacologiques et à leurs toxicités
différentes de celles des chimiothérapies conventionnelles. Les ITK du récepteur épidermique de facteur
de croissance épithélial (
Epidermal growth factor receptor
[EGFR]) ont apporté un bénéfice important
chez les patients porteurs d’une mutation de son domaine tyrosine kinase. La translocation du gène ALK
(Anaplastic Lymphoma Kinase)
définit un deuxième groupe de cancers bronchiques non à petites cellules
(CBNPC) sensibles à un ITK. L’Institut national du cancer finance actuellement en routine la recherche
de6anomalies moléculaires du tissu tumoral permettant de sélectionner les patients répondeurs
auxthérapies ciblées telles que les ITK. Tous les acteurs de l’oncologie thoracique se doivent donc d’acquérir
une meilleure connaissance de ces molécules.
Mots-clés
Cancer bronchique
Thérapies ciblées
Inhibiteurs de tyrosine
kinase
EGFR
ALK
Summary
Tyrosine kinase inhibitors (TKI)
entered since 2005 the current
practice in thoracic oncology,
and bound the oncologist to
adapt to their pharmacological
properties and toxicities which
are different from conventional
chemotherapies. For patients
harboring EGFR tyrosine kinase
mutations, EGFR TKIs brought
a considerable benefit. ALK
translocation defines a 2nd
group of NSCLC sensitive
to a TKI. Presently, the INCa
launched routine testing of
6 molecular abnormalities of
tumoral tissue, allowing selec-
tion of patients responders to
targeted therapies such as TKIs.
A better knowledge of these
therapies by all the actors of
thoracic oncology is therefore
warranted.
Keywords
Lung neoplasm
Targeted therapies
Tyrosine kinase inhibitors
EGFR
ALK
4 exons du domaine tyrosine kinase (exons 18 à 21).
Les mutations des exons 19 et 21, les plus fréquentes,
sont activatrices, tandis que celles de l’exon 20 sont
plus rares et communément associées à une résis-
tance aux ITK-EGFR (4).
Les ITK-EGFR ont d’abord obtenu leur AMM en
deuxième ou en troisième ligne après un traitement
à base de sels de platine testé dans des populations
non sélectionnées de patients métastatiques, mais
ayant néanmoins un profil de patients répondeurs
non fumeurs, de sexe féminin, d’origine asiatique
et atteints d’adénocarcinome
(5). Les études ont
ensuite porté sur des populations de patients mutés
ou sur des populations enrichies en patients mutés
(en les sélectionnant sur des critères cliniques) :
on y retrouve des survies sans progression (SSP)
sous ITK-EGFR comprises entre 9,2 et 13,1 mois,
versus 4,6 à 6,3 mois pour les différents doublets
de chimiothérapie testés (tableau II) [6]. La seule
étude européenne comparant la bichimiothérapie à
un ITK-EGFR chez des patients mutés EGFR (étude
EURTAC) retrouve des SSP conformes aux précédents
résultats (9,4 versus 5,2 mois ; HR = 0,42) et une
médiane de survie globale (SG) qui n’est pas atteinte
après un suivi médian de 19 mois (7).
Les ITK-EGFR sont désormais un standard dans la
prise en charge des patients porteurs d’une mutation
de l’EGFR, avec des AMM obtenues en 2009 pour le
géfitinib et en 2011 pour l’erlotinib.
Une nouvelle génération d’ITK-EGFR venant se lier
de manière irréversible avec l’EGFR est actuellement
en développement.
Tableau II. Essais de phase III comparant les ITK-EGFR de première génération à un doublet de chimiothérapie chez des
patients porteurs d’une mutation de l’EGFR.
Étude/population Effectif SSP en mois
TKI (IC95) vs chimio HR (IC95)Tx réponse
(%)
Tx contrôle
(%)
IPASS/asiatique
(Géfitinib vs carboplatine-paclitaxel)
261 ND
(pour la population mutée)
0,48 (0,36-0,64) 71,2 ND
WJTOG/asiatique
(Géfitinib vs cisplatine-docétaxel)
177 9,2 (8-13,9) vs 6,3 0,49 (0,34-0,71) ND 93
NEJ002/asiatique
(Géfitinib vs carboplatine-paclitaxel)
228 10,8 (ND) vs 5,4 0,3 (0,22-0,41) 73,7 89,5
OPTIMAL/asiatique
(Erlotinib vs carboplatine-gemcitabine)
165 13,1 (10,6-16,5) vs 4,6 0,16 (0,1-0,26) 82 96
EURTAC/caucasienne
(Erlotinib vs doublet avec cisplatine)
174 9,7 (8,4-12,3) vs 5,2 0,37 (0,25-0,54) 58 79
SSP : survie sans progression ; Tx contrôle : taux de contrôle de la maladie (réponses complètes + réponses partielles + maladies
stabilisées) ; Tx réponse : taux de réponse (réponses complètes + réponses partielles).
Tableau I. Principaux biomarqueurs en 2012 financés par l’Institut national du cancer et ITK commercialisés ou en cours
d’évaluation.
Biomarqueur Fréquence ITK en pratique quotidienne ITK en essai
EGFR 19/21 12 % Erlotinib/géfitinib (AMM) Afatinib (BIBW2992), dacomitinib (PF00299804)
EGFR T790M 3 % Afatinib (BIBW2992), dacomitinib (PF00299804) ;
nouveaux inhibiteurs spécifiques
EML4-ALK 4 % Crizotinib (ATU de cohorte)
KRas 20 % Inhibiteurs de Mek ou BRaf (sorafénib)
BRaf (V600E) 2 % Inhibiteur de BRaf : vémurafénib ; GSK2118436
Pi3K 1% Inhibiteur de Pi3K : BKM120
HER2 1% Lapatinib ; afatinib (BIBW2992)

THÉRAPEUTIQUE
La Lettre du Pharmacologue • Vol. 26 - n° 3 - juillet-août-septembre 2012 | 95
Inhibiteurs de tyrosine kinase
de l’ALK (ITK-ALK)
L’Anaplastic Lymphoma Kinase (ALK) est un récep-
teur transmembranaire à activité tyrosine kinase
impliqué dans la cancérogenèse. Décrit dans une
hémo pathie depuis les années 1990, c’est en 2007
que la fusion EML4-ALK est pour la première fois
identifiée comme étant impliquée dans le cancer
bronchique non à petites cellules (CBNPC) [8]. Cette
translocation se retrouve dans 5 à 6 % des adéno-
carcinomes bronchiques
(9) et touche préférentielle-
ment les hommes jeunes non ou peu fumeurs ayant
un type histologique mucineux (10). La présence
d’une translocation ALK exclut le plus souvent les
autres anomalies moléculaires connues (EGFR, KRas)
[9]. Les patients transloqués ALK ne répondent pas
aux ITK-EGFR, mais ils semblent mieux répondre au
pémétrexed que les patients non transloqués (11). En
2008 a débuté la phase I d’un nouvel ITK, le crizo-
tinib, développé initialement comme un anti-Met.
Sur les 1 500 patients analysés, on en a retrouvé
82 qui étaient transloqués ALK, dont 79 porteurs
d’un adénocarcinome et 1 porteur d’un carcinome
épidermoïde. On a observé des taux de réponses
importants, avec 57 % de réponses objectives et
33 % de maladies stabilisées. La SSP à 6 mois était
de 72 % (médiane non atteinte) [12]. La poursuite
de cette phase I, avec 119 patients inclus et un
recul médian de 11 mois, retrouve une médiane de
SSP de 10 mois et une médiane de survie globale
non atteinte. Aucun des patients répondeurs n’est
amplifié Met, et c’est bien par la voie ALK que le
crizotinib semble agir.
Étant donné la rapidité de développement du crizo-
tinib, il est difficile de connaître exactement sa place
dans la stratégie thérapeutique des CBNPC translo-
qués ALK. Néanmoins, il est actuellement testé en
phase III en première ligne, et il ne fait aucun doute
que tous les patients transloqués ALK devront rece-
voir du crizotinib durant l’évolution de leur maladie,
à l’instar des patients mutés EGFR devant recevoir
des ITK-EGFR.
Inhibiteurs de tyrosine kinase
ciblant les autres anomalies
moléculaires
Les voies Pi3K/Akt et Ras/Raf/Mek sont les 2 prin-
cipales voies à l’étude dans le CBNPC.
Les mutations de KRas se caractérisent par leur
fréquence (7,1 % des carcinomes épidermoïdes et
18,4% des adénocarcinomes [13]) et par la difficulté
à élaborer des stratégies d’inhibition efficaces. Une
mutation de KRas est responsable d’une diminu-
tion de la réponse aux ITK-EGFR dans le CBNPC,
qui passe de 20 % à moins de 3 % selon le carac-
tère sauvage ou muté du CBNPC (14). En effet, en
cas de mutation ponctuelle de KRas, les signaux de
prolifération cellulaire deviennent indépendants de
l’EGFR. La signalisation de KRas passe par les 2 voies
synergiques de Pi3K/Akt et Ras/Raf/Mek, dont la
double inhibition paraît logique (15). Actuellement,
l’inhibition de Mek est décevante dans le CBNPC
avancé (16), mais active sur les lignées cellulaires
mutées sur KRas (17), et doit donc être étudiée dans
une population sélectionnée porteuse d’une muta-
tion de KRas. Le sorafénib (TKI BRaf) a montré des
résultats intéressants chez les patients mutés KRas
(essai BATTLE) [18], sans que l’on puisse distinguer
son action anti-VEGF de son action sur la voie Ras/
Raf/Mek.
Après le succès prometteur du vémurafénib dans
le mélanome [19], les ITK-BRaf sont actuellement
testés sur les 3 % de patients porteurs d’une muta-
tion BRaf (essentiellement V600E, patients fumeurs),
pour l’instant en deuxième ligne de traitement (8).
Concernant la voie Pi3K/Akt, des ITK-Pi3K sont
actuellement en développement pour les patients
porteurs d’une mutation Pi3K ou PTEN.
Enfin, les mutations de HER2, représentant de 1 à
4 % des adénocarcinomes bronchiques, semblent
accessibles aux traitements anti-HER2 connus, ainsi
qu’à l’afatinib (20) et au lapatinib.
Pharmacologie des inhibiteurs
de tyrosine kinase
Les ITK agissent par compétition avec l’ATP au niveau
de la poche de fixation à l’ATP. Leur cible est donc
intracellulaire : soit sur la partie intramembranaire
des récepteurs à activité tyrosine kinase (EGFR), soit
cytosolique sur des protéines de fusion purement
intracellulaires (EML4-ALK ou BCR-ABL).
Ils ont tous une administration orale et des demi-vies
élevées, mais l’effet de la prise alimentaire varie d’un
ITK à l’autre. Leur volume de distribution est élevé et
leur liaison aux protéines plasmatiques est très forte
(> 90 %), avec un risque de variation de la fraction
libre du médicament en cas d’inter action médicamen-
teuse ou d’hypoalbuminémie. Les ITK sont métabo-
lisés par le cytochrome CYP3A4, avec une excrétion
majoritaire sous forme de métabolites retrouvés dans
les fèces. On retrouve une faible proportion d’ITK

THÉRAPEUTIQUE
96 | La Lettre du Pharmacologue • Vol. 26 - n° 3 - juillet-août-septembre 2012
Indication et utilisation desinhibiteurs de tyrosine kinase dans le cancer
bronchique nonà petites cellules
sous forme inchangée dans les urines. La fonction
rénale a donc peu d’influence sur les ITK, alors que
des insuffisances hépatiques, si elles sont sévères,
doivent entraîner une surveillance accrue.
Certains ITK ont des particularités métaboliques
qui méritent d’être connues. Ainsi, l’erlotinib est
également métabolisé par les CYP1A1 et 1A2, pour
lesquels le tabac est un inducteur enzymatique. Un
tabagisme actif est donc une cause de baisse d’effi-
cacité de l’erlotinib. On notera enfin que, du fait de
leurs caractéristiques respectives, l’erlotinib doit être
pris à distance d’un repas (au moins 1 heure avant
ou 2 heures après), tandis que le géfitinib peut être
pris au cours d’un repas.
Toxicité
Les ITK obligent à prendre un peu de recul sur la
classification des effets indésirables NCI CTCAE
(tableau III). Une toxicité de grade 3 durant 24 à
48 heures après une chimiothérapie conventionnelle
toutes les 3 semaines peut parfois être mieux tolérée
qu’une toxicité de grade 2 durant plusieurs mois
de traitement quotidien par un ITK. Les principales
toxicités liées à la prise d’ITK-EGFR sont digestives
et cutanées. L’apparition d’une sécheresse cutanée
sur laquelle va se développer un rash acnéiforme
est tellement fréquente qu’il est licite de proposer
systématiquement un traitement préventif, à base
de crème hydratante sur les zones sèches associée
à une tétracycline p.o. ou à un dermocorticoïde
pour l’éruption acnéiforme. Sur le plan digestif,
les ITK-EGFR entraînent également de fréquentes
diarrhées, ainsi qu’une cytolyse hépatique le plus
souvent asymptomatique et réversible. L’effet indé-
sirable le plus grave est la survenue de pneumo-
pathies interstitielles, potentiellement fatales, mais
heureusement rares.
Le crizotinib présente une toxicité visuelle n’entraî-
nant que rarement un arrêt du traitement. On note
fréquemment des éclairs lumineux décrits par les
patients, notamment lors du passage de l’ombre
à la lumière (12). Les toxicités les plus fréquentes
sont des nausées et des diarrhées de grade 1. Les
plus graves restent rares : une cytolyse hépatique
peut survenir dans les 2 premiers mois, ainsi que
l’apparition de pneumopathies interstitielles aiguës.
Résistance aux inhibiteurs
de tyrosine kinase
Même si les ITK-EGFR ont amélioré la SSP des
patients, ils se heurtent toujours au développement
de résistances.
La majorité de ces résistances se traduit par l’ap-
parition d’une seconde mutation sur le domaine
tyrosine kinase, ce qui va amoindrir l’efficacité de
l’ITK en diminuant son affinité pour la poche à ATP.
Ce mécanisme de résistance aux ITK-EGFR apparaît
avec la mutation T790M au niveau de l’exon 20 pour
les ITK-EGFR (21) et avec les mutations L1196M,
C1156Y et G1269A pour le crizotinib (22). Des ITK de
Tableau III. Principales toxicités liées à la prise d’ITK. Données issues des résumés des caractéristiques du produit (dictionnaire Vidal© et Agence nationale de
sécurité du médicament et des produits de santé), de l’étude IPASS pour le géfitinib, de l’étude EURTAC pour l’erlotinib, et de l’étude de phase I du crizotinib.
Toxicité Erlotinib Géfitinib Crizotinib
Tout grade (%) Grade 3/4 (%) Tout grade (%) Grade 3/4 (%) Tout grade (%) Grade 3/4 (%)
Hématologique Neutropénie 0 0 3,7 < 10 < 5
Cutanée Rash/acné 65-75 8-12 66 3 9 0
Xérose 12 0 24 0
Digestive
Diarrhées 48-52 5 46 4 43 0
Anorexie 30 0 22 1,5 17 0
Nausées
Vomissements
33
23
3
2
17
13
0,3
0,2
50
35
0
0
Cytolyse hépatique 3 2 9110-15 52
Asthénie 52 6-14 17 0,3 17 2
Pneumopathie interstitielle 1 1-2 22
Cardiaque Bradycardie
Allongement QTc
5
1
0
0
Troubles visuels 75-80 0
1 Asymptomatique et réversible.
2 Dont décès toxique.

THÉRAPEUTIQUE
La Lettre du Pharmacologue • Vol. 26 - n° 3 - juillet-août-septembre 2012 | 97
nouvelle génération sont actuellement développés
pour former des liaisons covalentes et irréversibles
avec le domaine tyrosine kinase, entraÎnant une inhi-
bition qui n’est alors plus compétitive. On citera en
particulier l’afatinib et le dacomitinib, qui semblent
avoir une efficacité supérieure à celle des ITK-EGFR
de première génération et pouvoir rattraper certains
de leurs échecs (23).
Par ailleurs, l’apparition d’amplifications du gène
cible est retrouvée dans certains cas de récidive
après un traitement par ITK-EGFR (21) ou par
ITK-ALK (22).
Enfin, la résistance est parfois acquise par la mise
en jeu de nouvelles voies de signalisation qui court-
circuitent l’anomalie motrice initialement ciblée.
On retrouve ainsi des amplifications de Met (récep-
teur à activité tyrosine kinase de l’EGF) visant à
rétablir l’activité de la voie Pi3K/Akt via la dimé-
risation avec HER (21). Met devient donc une cible
de choix, actuellement en essais thérapeutiques en
traitement de deuxième ligne (24).
Conclusion
L’Institut national du cancer finance actuellement en
France la réalisation de tests de biologie moléculaire
ciblant EGFR, KRas, BRaf, Pi3K, HER2, ainsi que la
translocation EML4-ALK. Ce programme de finance-
ment de 6 biomarqueurs dans le cadre d’un réseau
recouvrant tout le territoire fait du CBNPC le cancer
le mieux doté en termes de biologie moléculaire.
La mise en évidence de ces anomalies génétiques
justifie l’utilisation de traitements ciblés qui ont le
plus souvent une activité inhibitrice de la fonction
tyrosine kinase de ces protéines. Certains sont parfai-
tement validés, tels que les ITK-EGFR, d’autres sont
en passe de l’être, tels que le crizotinib, et la plupart
sont en cours d’investigation.
Cette nouvelle approche thérapeutique justifie une
meilleure connaissance de leur mécanisme d’action,
de leur efficacité, mais aussi de leur toxicité et des
mécanismes de résistance par tous les acteurs de
la prise en charge de ces patients. ■
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next
generation. Cell 2011;144:646-74.
2. Weinstein IB, Joe AK. Mechanisms of disease: Oncogene
addiction – a rationale for molecular targeting in cancer
therapy. Nat Clin Pract Oncol 2006;3:448-57.
3. Kris MG, Johnson D, Kwiatkowski DJ et al. Identification
of driver mutations in tumor specimens from 1,000 patients
with lung adenocarcinoma: The NCI’s Lung Cancer Mutation
Consortium. ASCO 2011. Abstract CRA 7506.
4. Sharma SV, Bell DW, Settleman J, Haber DA. Epidermal
growth factor receptor mutations in lung cancer. Nat Rev
Cancer 2007;7:169-81.
5. Shepherd FA, Rodrigues Pereira J, Ciuleanu TS et al. Erlo-
tinib in previously treated non-small-cell lung cancer. N
Engl J Med 2005;353:123-32.
6. Mok TS, Wu YL, Thongprasert S et al. Gefitinib or carbo-
platin-paclitaxel in pulmonary adenocarcinoma. N Engl J
Med 2009;361:947-57.
7. Rosell R, Carcereny E, Gervais R et al. Erlotinib versus
standard chemotherapy as first-line treatment for Euro-
pean patients with advanced EGFR mutation-positive non-
small-cell lung cancer (EURTAC): a multicentre, open-label,
randomised phase 3 trial. Lancet Oncol 2012;13:239-46.
8. Soda M, Choi YL, Enomoto M et al. Identification of the
transforming EML4-ALK fusion gene in non-small-cell lung
cancer. Nature 2007;448:561-6.
9. Kris MG. Identification of driver mutations in tumor speci-
mens from 1,000 patients with lung adenocarcinoma: The
NCI’s Lung Cancer Mutation Consortium (LCMC). ASCO
Meeting Abstracts 2011;29(15): CRA7506. 2011.
10. Inamura K, Takeuchi K, Togashi Y et al. EML4-ALK lung
cancers are characterized by rare other mutations, a TTF-1
cell lineage, an acinar histology, and young onset. Mod
Pathol 2009;22:508-15.
11. Lee JO, Kim TM, Lee SH et al. Anaplastic lymphoma
kinase translocation: a predictive biomarker of pemetrexed
in patients with non-small-cell lung cancer. J Thorac Oncol
2011;6:1474-80.
12. Kwak EL, Bang YJ, Camidge DR et al. Anaplastic
lymphoma kinase inhibition in non-small-cell lung cancer.
N Engl J Med 2010;363:1693-703.
13. Mascaux C, Iannino N, Martin B et al. The role of RAS
oncogene in survival of patients with lung cancer: a syste-
matic review of the literature with meta-analysis. Br J Cancer
2005;92:131-9.
14. Raponi M, Winkler H, Dracopoli NC. KRAS mutations
predict response to EGFR inhibitors. Curr Opin Pharmacol
2008;8:413-8.
15. Sos ML, Fischer S, Ullrich R et al. Identifying genotype-
dependent efficacy of single and combined PI3K- and
MAPK-pathway inhibition in cancer. Proc Natl Acad Sci
USA 2009;106:18351-6.
16. Hainsworth JD, Cebotaru CL, Kanarev V et al. A phase
II, open-label, randomized study to assess the efficacy and
safety of AZD6244 (ARRY-142886) versus pemetrexed in
patients with non-small-cell lung cancer who have failed
one or two prior chemotherapeutic regimens. J Thorac Oncol
2010;5:1630-6.
17. Garon EB, Finn RS, Hosmer W et al. Identification of
common predictive markers of in vitro response to the Mek
inhibitor selumetinib (AZD6244; ARRY-142886) in human
breast cancer and non-small-cell lung cancer cell lines. Mol
Cancer Ther 2010;9:1985-94.
18. Kim ES, Herbst RS, Wistuba II et al. The BATTLE Trial: perso-
nalizing therapy for lung cancer. Cancer Discov 2011;1:44-53.
19. Chapman PB, Hauschild A, Robert C et al. Improved
survival with vemurafenib in melanoma with BRAF V600E
mutation. N Engl J Med 2011;364:2507-16.
20. De Grève J, Teugels E, Geers C et al. Clinical activity of
afatinib (BIBW 2992) in patients with lung adenocarcinoma
with mutations in the kinase domain of HER2/neu. Lung
Cancer 2012;76:123-7.
21. Sequist LV, Waltman BA, Dias-Santagata D et al.
Genotypic and histological evolution of lung cancers
acquiring resistance to EGFR inhibitors. Sci Transl Med
2011;3:75ra26.
22. Doebele RC, Pilling AB, Aisner DL et al. Mechanisms of
resistance to crizotinib in patients with ALK gene rearranged
non-small-cell lung cancer. Clin Cancer Res 2012;18:1472-82.
23. Miller VA, Hirsh V, Cadranel J et al. Afatinib versus
placebo for patients with advanced, metastatic non-small-
cell lung cancer after failure of erlotinib, gefitinib, or both,
and one or two lines of chemotherapy (LUX-Lung 1): a phase
2b/3 randomised trial. Lancet Oncol 2012;13:528-38.
24. Wang W, Li Q, Takeuchi S et al. Met kinase inhibitor
E7050 reverses three different mechanisms of hepatocyte
growth factor-induced tyrosine kinase inhibitor resistance
in EGFR mutant lung cancer. Clin Cancer Res 2012;18:
1663-71.
Références bibliographiques
1
/
5
100%