Antiagregants-plaquettaires (pdf)

UE 10– Tissu sanguin
Guerin-Dubourg
Date : 15/02/2016 Plage horaire : 14-16h
Promo : P2 Enseignant : Guerin-Dubourg
Ronéistes :
LEVASSEUR Benjamin
BORDIER Alexandre
LEGRAND Jean-Baptiste
SAINT-ALME Sébastien
Pharmacologie de la pathologie thrombo-embolique
I. Anticoagulant
1. Les héparines
2. Héparinoïdes
3. Nouveaux anticoagulants
4. Les AVK
II. Antiagrégants plaquettaires
1. Inhibiteur de l’amplification de TXA2
2. Potentialisateurs de l’AMPc
3. Les inhibiteurs du récepteur de l’ADP P2Y12
4. Les inhibiteurs du GPIIbIIIa activé

Introduction
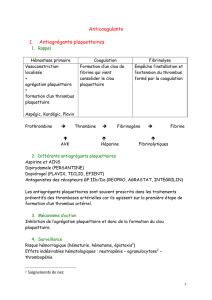
Petit rappel sur la coagulation :
Les pathologies thromboemboliques sont des pathologies qui sont liées à des troubles de la coagulation.
Il y a plusieurs mécanismes de coagulation : _ l’hémostase primaire (vasoconstriction et l'adhésion
plaquettaire au vaisseau sanguin lésé et en l'agrégation plaquettaire)
_l’hémostase secondaire ou coagulation (formation du
caillot/agrégat plaquettaire en lui-même)
Le fibrinogène et les facteurs certain facteurs de coagulation sont contenus dans le sérum
Quand on parle de caillot de fibrine, c’est en fait une gélification du sérum (qui est normalement quelque
chose d’assez liquide), le thrombus étant l’agrégat plaquettaire qui se forme autour du caillot de fibrine.
Les maladies thromboemboliques correspondent à l’occlusion d’un vaisseau par un amas de cellules
(essentiellement des plaquettes) et/ou de fibrine. Les facteurs favorisants sont la stase, l’âge, les syndromes
d’hypercoagulabilité (risque familial de thrombophilie), l’hypertension, l’hyperlipémie, les
lésions/dysfonction de l’endothélium (relargage de facteurs tissulaires => activation de la coagulation),
toutes les formes de cancer qui vont avoir tendance à activer la coagulation de façons assez anarchiques.
On va avoir deux types de thromboses : veineuses avec trouble de la coagulation au niveau du sérum et
artérielles avec agrégation plaquettaire et formation de thrombus (syndrome coronarien, chirurgie cardiaque).
Donc pour les thromboses veineuses on utilisera des anticoagulants (héparine, nouveaux anticoagulants,
AVK) et pour les artérielles on utilise plutôt des anti-agrégants plaquettaires. On ne traitera pas des
thrombolytiques qui servent aux traitements curatifs dans l’urgence (lors d’un AVC ischémique par exemple).
Les TVP peuvent amener à un déplacement du caillot et donner des œdèmes aigue du poumon ou des
ischémies au niveau du SNC.
Le mécanisme physiopathologique classique (mais ce n’est pas le seul) de la thrombose artérielle est la plaque
d’athérome. On parle d’athérosclérose, mécanisme inflammatoire au niveau des parois vasculaire qui rigidifie
les vaisseaux. On a une rupture de cette plaque d’athérome qui amène à un relargage de facteurs de formation
d’un agrégat de fibrine (un peu), mais surtout de formation de l’agrégat plaquettaire comme le calcium. Cela
amène à la formation d’un thrombus avec réseau de fibrine et plaquettes qui mènent à une occlusion, ce qui
entraine des pathologies comme l’IDM, l’AVC ou les artérites des MI.
On va parler dans ce cours de deux grandes familles d’anti-thrombotique :
_ Les antiagrégant plaquettaire qui vont avoir vocation à prévenir la formation des thrombus artériel
_ Les anticoagulant qui vont limiter plutôt l’apparition de thrombose veineuse
On ne parlera par contre pas dans ce cours des thrombolytique/fibrinolytique, qui sont des médicaments
d’urgence qui visent à pulvériser/détruire un caillot déjà formé (fibrinolyse). Ce sont des médicaments à
utiliser avec énormément de prudence.
La coagulation est un mécanisme de cascade enzymatique (du facteur XIII jusqu’au facteur II) :
On a une activation successive des facteurs de la coagulation (thrombine, fibrinogène, fibrine). Les éléments
les plus importants sont le facteur II et X, ce sont des cibles pharmacologiques privilégiées, et plus on agit en
amont de la cascade de coagulation, plus on aura un effet puissant. On a fait des tentatives pour agir plus haut,
mais les risques hémorragiques devenaient trop importants. Le facteur II va être activé par le facteur V et
facteur Xa (X activé) pour donner le facteur IIa (appelé thrombine) qui est capable de transformer le
fibrinogène en fibrine (capacité de coagulation par création d’un réseau de fibrine).
Les anticoagulants sont des médicaments qui vont agir sur cette cascade enzymatique, ce qui va in fine inhiber
la transformation du fibrinogène en fibrine.
A propos du test biologique, et pour résumer grossièrement : la TCA et la TCK explorent l’activité de
l’héparine alors que les INR explorent l’activité des AVK

I. Anticoagulants
1. Héparines
Les héparines font partie des glycosaminoglycanes (répétitions d’unités dissacharidiques) et comportent des
structures pentasaccharidiques qui portent la principal activité anticoagulante. Elles sont présentes au niveau
de l’endothélium et agissent en lien avec l’antithrombine en augmentant sa capacité anticoagulante, c’est pour
cela qu’on dit qu’elle a une activité indirecte : son activité est porté par l’antithrombine, qu’elle potentialise.
Les Héparine se donne forcément en sous cutané car étant constitué de GAG elles se digèrent.
On distingue les HNF (héparines non fractionnées avec PM moyen 15 000) qu’on extrait chez le porc,
et les HBPM (héparines de bas poids moléculaire avec PM < 8000) qu’on obtient après fractionnement
chimique ou digestion enzymatique. Ces deux héparines portent les structures pentasaccharidiques mais se
distinguent par les chaînes. Les HNF aux longues chaînes vont avoir une activité anticoagulante
supplémentaire non désirée car difficile à contrôler. Les HBPM sont donc un peu moins puissantes que les
HNF, mais sont surtout plus facile à maîtriser. Les HBPM sont beaucoup plus courtes.
Les héparines agissent sur un facteur anticoagulant naturel qui est l’anti-thrombine. Elle agit sur la thrombine
(facteur IIa) mais aussi sur le facteur Xa. L’anti-thrombine est un facteur physiologique de régulation de la
coagulation qui, liée à la thrombine, empêche son activité et donc la de formation de thrombine et de fibrine.
/!\ Retenir que l’anti-thrombine agit principalement sur le facteur Xa !
Le facteur Xa couplé au Va active le IIa. L’héparine va potentialiser l’action de l’anti-thrombine. L’héparine
a donc majoritairement une activité anti-Xa indirecte (et minoritairement une activité anti-IIa indirecte non
désirée).
L’anti-thrombine a d’abord un effet sur le Xa, elle est capable de lier le IIa. Ce qu’il faut savoir, c’est que
l’activité anti-Xa indirecte de l’anti-thrombine est potentialisée par le pentasaccharide de l’héparine. Mais si
on a une longue chaine de glycosaminoglycanes sur l’héparine, on va également potentialiser l’effet anti-IIa
indirecte de l’antithrombine, l’activité anti-IIa de l’antithrombine, bien qu’existante physiologiquement, étant
essentiellement présente quand elle est liée à l’héparine, celle-ci permettant au IIa de se lier à l’antithrombine.
En fonction de la taille des glycosaminoglycanes, du poids moléculaire de l’héparine, on aura une activité
anti-IIa plus ou moins importante.

HNF: activité anti Xa et IIa indirecte
importante.
HBPM: plus leur PM est bas, plus
l’activité anti-IIa va diminuer donc
elles auront majoritairement une
activité anti-Xa
S’il ne nous reste que le
pentasaccharide, c’est ce qu’on appelle
les héparinoïdes, on a une activité anti-
Xa seule indirecte.
Plus le rapport anti-Xa/ anti-IIa est élevé,
plus il aura une activité anti-Xa. Plus on
baisse dans les PM, plus le rapport anti
Xa/anti IIa augmente, donc une activité
anti-Xa prépondérante.
Le problème de l’activité anti-IIa est
qu’elle est puissante et donc dure à
maitriser, avec de gros risque d’accident
iatrogène. C’est aussi pour ça que les
HNF ont souvent une activité plus
importante que les HBPM.
Les HNF ont une forte variabilité inter-
individuelle du fait de leur forte activité
anti-IIa (qu’on a du mal à gérer) : il faut
donc adapter la dose en fonction des
résultats biologiques.
Il faut aussi faire une surveillance quotidienne (TCA [temps de céphaline activée ou PTT], TCK, taux de
plaquettes). Il y a 2 à 3 injections par jour, l’élimination se fait par le système réticulo-endothélial ou rénal.
La surveillance du taux de plaquette est nécessaire car celles-ci ont tendance à diminuer avec l’héparine, ce
qui entraine un risque iatrogène.
Les HBPM ont une bonne prédictibilité (moins de variabilités inter-individuelles), il y a donc une dose fixe
en fonction du poids et pas besoin de surveillance biologique sauf chez les IR où on peut doser l’activité anti-
Xa (aussi appelé héparinémie – bien que l’on ne dose pas l’héparine mais bien l’activité anti-Xa) et où il y a
un risque moindre de thrombopénies induite à l’héparine (TIH). Une à deux injections par jour.
Les HNF et les HBPM ont une élimination rénale essentiellement.
Le TCA exploite principalement la voie endogène de la coagulation. Les HNF ou héparines standards vont
avoir un effet sur le TCA. Ce qui va changer un INR, ce sont les anti-vitamines K avec un retentissement sur
le TCA. Les HBPM agissent très peu sur le TCA.
On va faire la surveillance des plaquettes car il peut y avoir une thrombopénie (effet indésirable grave)
induite à l’héparine.
TIH : Thrombopénie immuno-allergique induite à l’héparine
A un patient sous héparine on donne un médicament anticoagulant : il peut y avoir une thrombopénie; risque
hémorragique important.

Les deux sont d’origine extractive et ont une action rapide voire immédiate, même si les HNF sont un peu
plus rapides car administrées en IV alors que les HBPM le sont par voie sous-cutanée. On prévient la formation
de caillot.
Bonus pour les QCM : les héparines, au contraire des AVK, ont un effet in vitro : si on met des héparines dans
un tube de sang il y aura une activité anticoagulante, ce qui n’est pas le cas avec les AVK.
Les indications : les maladies thrombo-emboliques veineuses ou artérielles. Les HNF ont parfois un rôle
curatif, mais ce sont essentiellement des médicaments utilisés à but préventif. Les HBPM servent au traitement
préventif dans la prophylaxie des pathologies thromboemboliques veineuses. D’abord on casse le caillot avec
un thrombolytique, puis on donne de l’héparine en préventif (en dose de charge au départ), ce qui empêche la
formation de nouveaux caillots. C’est essentiellement de la prévention.
Les précautions d’emploi : IH ou IR, HTA sévère – risque vasculaire important, sujets âgés, risques d’ulcère
(car risque hémorragique important).
Les contre-indications : retenir que les médicaments qui empêchent la coagulation ou l’agrégation
plaquettaire seront contre-indiqués.
- Les actes invasifs tels que les injections intra-musculaire, intra-artérielle, intra-articulaire, infiltration
sympathique car on a un risque d’hémorragie importante.
- Les associations déconseillées : AINS (qui ont un effet sur les prostaglandines qui ont elle-même des effets
antalgique, antipyrétique et antiagrégant) et salicylés, ticlopidine (= antiagrégants plaquettaire)
- Les associations avec prudence : AVK, corticoïdes, thrombolytique (médicaments d’urgence)
- Précaution d’emploi avec les patients qui ont un risque hémorragique
Lorsqu’on a un patient qui fait une thrombose veineuse profonde, on va le traiter : dose de charge,
thrombolyse, HNF souvent pour éviter qu’il refasse des caillots. A l’hôpital, en observation, on passe aux
HBPM, plus faciles à utiliser. Très rapidement, avant sa sortie, on fait un relais héparine/AVK.
Tous les anti-agrégants plaquettaires sont à déconseiller en association avec les héparines ou à utiliser avec
prudence.
Effets Indésirables
• Risque hémorragique : on a un surdosage, le patient peut saigner. Le moindre hématome peut se
transformer en hémorragie.
⇨ Antidote : Sulfate de protamine (plutôt au bloc)
• Réactions allergiques, éruptions cutanées au point d’injection
• Augmentation des transaminases (HNF) (via un effet de toxicité hépatique, généralement pas très
préoccupant)
• Thrombopénies induite à l’héparine(TIH)
La TIH est une thrombopénie très caractéristique qui est précoces et modérées, ça arrive au moment de
l’injection, le lendemain on a une baisse de nos plaquettes 10 000 à 20 000/ 200 000, ce n’est pas
significatif mais si dans les jours qui suivent le nombre des plaquettes continue à baisser, on aura une
thrombopénie induite à l’héparine. La meilleure façon de faire un diagnostic d’une TIH est d’arrêter le
traitement à l’héparine et de voir si le nombre de plaquettes augmente.
L’une des caractéristique de la TIH, et qui permet de faire le diagnostic différentiel avec d’autre type de
thrombopénie est qu’on a une thrombopénie sans signe hémorragique associé comme un purpura, on aura
au contraire plutôt des signes de thrombose.
En effet, dans les TIH, ce n’est pas vraiment le nombre de plaquette qui chute mais c’est plutôt qu’on fait
des caillots, tellement que quand on fait la NFS le nombre de plaquette semble chuter car on arrive plus
à les doser correctement.
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
6
7
8
9
10
11
12
13
14
15
16
17
18
19
1
/
19
100%