Etude génétique du cycle cellulaire et de sa régulation Introduction

Etude génétique du cycle cellulaire et de sa régulation
Introduction.
Comment a-t-on pu établir le cycle cellulaire de cette espèce ?
Chez tous les organismes : il y a des étapes faciles a observées comme la division cellulaire.
Chez les eucaryotes supérieurs, la mitose est également facilement observable.
Les populations cellulaires sont asynchrones : toutes les cellules ne se divisent pas en même
temps : techniques spéciales pour les synchroniser.
Estimation de la durée de la phase S.
On fait un marquage de l’ADN néo-synthétisé avec un marqueur appelé BrdU. Puis on
fixe immédiatement les cellules et on rajoute des Ac anti-BdrU. Ainsi, on ne marque que les
cellules qui sont à un moment précis de leur cycle et qui fixe donc ce BrdU. Pour les cellules
de mammifères, la phase S représente environ 1/3 du cycle.
Mais ou se situe exactement cette phase S dans un cycle ?
Estimation de la phase G2.
On rajoute du BrdU pendant différents temps, jusqu’à que l’on ne voit plus de
marquage. Cela marque la durée entre la fin de la phase S et le début de la mitose.
Analyse du cycle cellulaire par iodure de propidium (IP). [Il s’agit d’un agent intercalant : la
quantité fixée est directement liée à la quantité d’ADN.]
Le nocodazole est un poison du fuseau mitotique : les chromosomes ne peuvent pas migrer
aux pôles.
Les hépatocytes ont un contenu en ADN de type G1. Ils ne se divisent plus en temps normal.
Mais la morphologie des hépatocytes en G1 quiescent sont différents de ceux en G1
cyclant. : on a alors appelé ce stage G0.

1948 : premières cultures de cellules de mammifères. Quelques années plus tard, on est
capable de faire fusionner des cellules de mammifères ce qui donne des hétérocaryons. Les
cellules qui arrivent à survivre à la fusion et à la culture arrivent à un stade d’hybrides
somatiques (4n ADN).
Que se passe t’il si les cellules fusionnent ne sont pas au même stade cellulaire ?
G1 avec G2 : G1 fait ses G1, S, G2 pendant que G2 attend puis les deux cellules rentrent en
mitose.
G1 avec S : G1 écourte son S et rattrape l’autre puis les deux cellules rentrent en mitose.
Il existe donc des protéines régulatrices.
On garde toujours le même ordre des phases.
En G1 et S, il doit y avoir des inhibiteurs de rentrée en mitose pour le noyau de G2.
En S, il doit y avoir des activateurs de rentrée en S pour les cellules G1 et des inhibiteurs de
rentrée en S pour le noyau de G2.
Phénomènes hautement régulés.
Une fusion d’une cellule en mitose avec n’importe qu’elle cellule aux stades G1, S, G2
provoque une condensation des chromosomes (en G1 : chromosomes à une chromatide) (en
G2, ca se passe bien) (en S : pas viable).
Il existe un activateur de rentrée en M très puissant qui permet dans ce cas uniquement
d’inverser l’ordre des phases : c’est le MPF (mitosis promotting factor). On peut faire
condenser des chromosomes humains avec un noyau mitotique de souris, drosophile…etc.et
réciproquement.
Le MPF est très conservé.
Caractéristiques de S.cerevisae et S.pombe.
Génome haploïde : 17,5Mbases.
Cycle cellulaire chez S.cerevisae.
La cellule qui commence a présenté un bourgeon comme sa phase S. C’est un moyen
physique de reconnaitre les cellules qui rentrent dans ce stade. (Erreur sur le schéma du
polycopier).
Il s’agit d’un organisme haplo-diplo-biontique.

-haploïde : utile pour avoir des mutants récessifs
-diploïde : utile pour avoir des mutants dominants.
Les cellules de type A sécrètent un peptide A qui est capable de bloquer les cellules α au
stade Start, ainsi que les cellules de type α sécrètent un peptide α qui est capable de bloquer
les cellules A au stade Start. Les cellules sont donc au même stade avant de fusionner.
Mutants de cycles conditionnels (cdc) Prix Nobel 2001.
On va passer par des mutants thermosensibles (le phénotype de thermosensibilité
n’est pas forcement la létalité). C’est la protéine qui sera thermosensible. La protéine
sauvage résistera jusqu’à x degrés alors que la mutante y sera instable. On utilise en général
3 températures chez la levure :
-11°C : la levure se divise de façon normale (si <, alors on n’observe que peu de
divisions)
-36°C
-23°C
11°C
23°C
36°C
WT
+
+
+
Mutants TS
+
?
-
Mutants CS
-
?
+
Sélections de mutants létaux thermosensibles (TS) indépendants.
=>plusieurs mutants sont créés. On met en culture pour obtenir une large palette de
mutants différents. On ne veut surtout pas que deux mutants provenant de la même cellule
mère soit créés.
On commence à faire une culture à température non permissive pour éviter des mutants
déjà présents pour la protéine recherchée. Les mutants, qui n’ont pas le temps de se diviser,
ne tombe que dans un seul tube donné lors de l’élution. (On se dépêche de faire des aligots
pour éviter que le mutant ne se divise).
On met ensuite en culture sur un milieu riche pour éviter les parasitages des mutations qui
auraient été induites partout ailleurs (ex : mutants des voies métaboliques…)
Analyse génétique : procédure générale.
Combien de gènes sont touchés dans ces mutants ?
On étale les cellules sur un milieu Ura- Leu- parce que les diploïdes n’aient pas besoin d’Ura
et Leu pour vivre (Ura et Leu sont dominants).
Cependant, nos levures sont de même sexe, donc il va falloir créer soit des levures A ou des
levures α. On va faire passer la méiose aux levures pour les faire sporuler. Pour récupérer
les bonnes spores, et éviter les problèmes de probabilité de récupérer les bons mutants, on
va faire pousser les levures sur différents milieux sous différentes conditions. On récupère
alors cdc x, Ura3, Leu2, α/A. Il nous reste à savoir si la souche et A ou α ?
=>on fait des croisements arbitrairement avec des souches de références dont le sexe
est connu. On fait pousser sur un milieu sans Ura ni Leu pour que seules les cellules diploïdes
poussent. Si ca pousse, on verra l’apparition de colonies a l’intersection des stries sur la
boite de Pétri.
Si muté dans le même gène : cdc x
𝑐𝑑𝑐 𝑦 Si muté dans un gène différent : cdc x
𝑐𝑑𝑐 𝑋 cdc y
𝑐𝑑𝑐 𝑌

Résultats finaux : un panel de 32 gènes ont été identifiés.
Analyse fine des phénotypes.
On repère les cellules individuelles qui ont été étalées sur la boite et on regarde la forme de
ces cellules. On met en culture en température non permissive. Ils voient dans les deux types
de mutants des cellules avec leurs bourgeons mais dans d’autres cas il y a deux cellules avec
leurs bourgeons.
Certaines cellules se sont arrêtées en G2/M du premier cycle (1cellule avec un gros
bourgeon)
D’autres sont rentrées dans un cycle et se sont arrêtées dans le cycle suivant en
G2/M.
Cela permet de définir le point d’exécution.
Pour cdc7 : toutes les cellules sans bourgeons se sont arrêtées dans le 1er cycle et
toutes les cellules ayant un bourgeon se sont arrêtées dans le 2e cycle.
La protéine n’est importante que dans un moment précis du cycle. Si on porte à
température non permissives les cellules qui ont besoin de la protéine, alors il y aura
un blocage. Apres G1/S, la protéine n’est plus nécessaire.
Pour cdc8, son point d’exécution est en fin de S.
On peut donc avoir les deux mutants qui ont des phénotypes finaux similaires mais avec des
points d’exécutions différents.
Les cdc7 et cdc8 ont un faux phénotype terminal de type G1/S : il y a un bourgeon avec entre
les deux le noyau mais celui-ci n’a pas été répliqué. La cellule reste à 1n ADN après analyse
par la technique de FACS.
Puis, dans une autre expérience, on le laisse à température permissive jusqu’à ce qu’elles
passent S, et ensuite a température non permissive. Les cdc8 arrêtent leurs phases S
directement, alors que les cdc7 continuent comme a température permissive. Cdc7 est une
protéine d’initiation alors que cdc8 est une protéine d’élongation.
Le cycle de l’ADN est finalement indépendant de la morphogénèse.

Sur le polycopier : ++ signifie que la cellule est capable de faire plusieurs fois l’étape.
Ex : cdc24 mutant : abouti a une cellule polynuclée qui va mourir.
Dans une 2e série d’expériences, on ne part pas de 400 souches mais de 5000. On ne
découvre alors que 3 groupes de complémentations en plus.
Raison :
-certaines protéines ne se prêtent pas aux mutations.
-plusieurs gènes codent pour la même protéine ce qui empêche de voir les mutants.
(Des mutants sortent à plus basse fréquences que d’autres.)
Le couplage peut être mécanique (le produit d’une étape permet le passage à une
autre étape) ou moléculaire (des protéines régulatrice sont mises en jeu.)
On peut inhiber la ribonucléotides réductase par une molécule : l’hydroxyurée (HU). Par
manque de substrat, les polymérases vont ralentir et la phase S sera effacée (ou retardée).
Après traitement des cellules par l’HU, on observe de grosses cellules (car leurs cycles est
allongé : phase S quasiment doublée).
Est-ce qu’il existe des mutations qui abolissent ce couplage synthèse/mitose ?
Oui ! Cdc2-3wee chez S.pombe, cdc28 chez S.cerevisae.
C’est un mutant gain de fonction avec une protéine très active. Presque pas de G2 avant
pour rentrer en mitose plus vite a cause de cette kinase très active.
Avec le HU, les cellules rentrent en mitose avant la fin de la phase S. Le noyau est coupé par
le septum : létal. Cette mutation découple la phase S de la phase M.
C’est bien une régulation moléculaire qui fait ce couplage. La protéine cdc2 est
directement impliquée dans cette régulation.
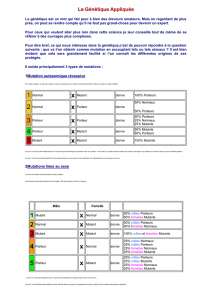
Exemple : cdc17 cdc2+ --------------X-----------------cdc17+ cdc2-3w
Phénotype TS non TS /non létal
Point d’initiation : S Petites cellules. Mutation
dominante Phénotype final G2/M
Cdc17 cdc2+
------- --------
Cdc17+ cdc2-3w
Après méiose : spores :
cdc17 cdc2+
cdc17+ cdc2-3w
 6
7
8
9
10
11
12
13
14
6
7
8
9
10
11
12
13
14
1
/
14
100%