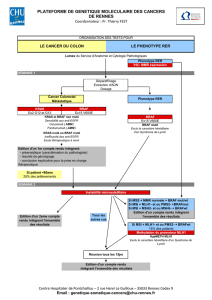
de la culture cellulaire au modèle animal

L'histiocytose Langerhansienne
et le polyomavirus de Merkel :
de la culture cellulaire au modèle animal
Ma. Victoria Ayala
Victor Chaumeton
Léonid Velikovsky
Sous l’encadrement du Pr. Laurence Nieto
M2P EGPR
2015 - 2016


Remerciement
En tout premier lieu, nous souhaitons vivement remercier notre professeur encadrante, Laurence,
qui a su nous guider, avec toute la pédagogie dont elle est capable, pour la réalisation de ce projet.
Nous saluons aussi sa patience (il en a fallu beaucoup), et son accessibilité. Son recul dans le domaine
de la recherche nous a été indispensable au cours de la mise en place de ce travail (ou jeu ?).
Nous remercions Laurent, pour nous avoir permis de suivre le cursus d’expression génique et de
protéines recombinantes, ainsi que pour sa vision de l’enseignement, à notre sens d’une qualité rare,
et pour les nombreuses discussions qui découlaient de ses interventions.
Nous tenons à remercier Remy pour ses conseils et ses éclaircissements, particulièrement au sujet
des maladies chroniques inflammatoires.
Nous souhaitons également remercier le reste de l’équipe pédagogique, Pascal, Samuel, Eric,
François, Vincent et Sylvie. Ensemble, ils ont participé à notre épanouissement intellectuel et
scientifique.
Enfin, nous remercions vivement la promotion n°16 du M2P EGPR, pour la qualité du travail fourni et
pour la bonne humeur régnant dans notre petite salle.


Abréviations
CCL20: chimiokine CCL20
DC: dendritic cell
Gluc : luciférase Gaussia
GM-CSF : facteur de stimulation de colonies de granulocytes et de macrophages
HL-MS : HL multi-systémique
HL-RO - : HL dans les organes non à risque élevé
HL-RO + : HL dans les organes à risque élevé
HL-SS : HL à système unique
hPBMC : cellules mononucléaires humaines du sang périphérique
IFN- γ : interféron-γ
IL : interleukine-1
IL-17A : interleukine 17A
IL-1R : récepteur de l’interleukine 1
LC : cellules de Langerhans
HL : Histiocytose Langerhansienne
MCC : Merkel Cell Carcinoma
MCMV : cytomégalovirus murin
MCPyV/MCV : Merkel cell polyomavirus
MGCs : cellules géantes multinuclées
MoDC : cellules dendritiques dérivées de monocytes
NCRR: non-coding regulatory region
PAMP: pathogen-associated microbia pattern
Pfu : unités formant des plages
Tg : Transgenic
Th17: lymphocytes T auxiliaires 17
TLR: Toll-like receptor
TNF-α : facteur de nécrose tumorale α
TRAP : phosphatase acide tartrate-résistante
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
1
/
58
100%