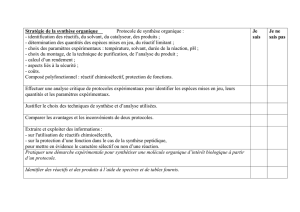
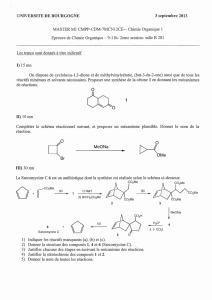
K - ESI

147
4.2.3 Demi-vies
• L’une des indications les plus utiles pour évaluer
la vitesse d’une réaction chimique d’ordre un est la
demi-vie, t1/2, d’un réactif, c’est-à-dire le temps
requis pour que la conc’n du réactif tombe à la
moitié de sa valeur initiale.
• On peut trouver le demi-vie d’une espèce A qui se
décompose par une réaction d’ordre un:
[A]=1
2[A]
o
et t = t1/2
ln [A]
[A]
0=−kt
(réaction d’ordre un)
ln1
2[A]
o
[A]
o=−kt12
kt12=−ln1
2=ln2
t12=ln2
k=0.693
k

148
• Donc, pour une
réaction d’ordre
un, la demi-vie
d’un réactif est
indépendante de
sa concentration
initiale.
• Par conséquence,
quelle que soit la
valeur réelle de
[A] à une étape
arbitraire de la
réaction, la conc’n
tombe à 1/2 [A]
après un intervalle
de 0.693/k.
• Pour des réactions d’ordre deux, la demi-vie
dépend de la conc’n initiale du réactif. En
générale,
t12∝1
[A]o
n−1
où n est l’ordre de la réaction.

149
4.3 Réactions d’ordre deux
• Pour la réaction générale: A Õ P
• La loi de vitesse est: vitesse=
−d[A]
dt =k[A]
2
où k (M-1s-1) est la constante de vitesse d’ordre
deux.
• En réarrangeant les variables et en intégrant cette
loi de vitesse, on obtient:
d[A]
[A]
2
[A]o
[A]
∫ =− kdt
0
t
∫
1
[A]=1
[A]
o+kt
ou
[A]=[A]
o
1+kt[A]
o

150
1
[A]
t
pente = k

151
4.4 Détermination de la loi de vitesse
4.4.1 Méthode d’isolement:
• On considère que tous les réactifs sauf un sont
présents en large excès. On peut dire avec une
bonne approximation que la concentration du
réactif en excès reste constante tout au long de la
réaction:
• Exemple: vitesse = k[A][B]2 et B est en excès
• On peut considérer que [B] est voisine de sa
valeur initiale [B]o et écrire
vitesse = k’[A] (pseudo-ordre un) avec k’=
k[B]o
2
• Si au contraire [A] était largement excédentaire,
on simplifierait la loi de vitesse
vitesse = k”[B]2 (pseudo-ordre deux)
avec k’’=
k[A]
o
• On peut trouver comment la vitesse dépend de
chaque réactif en isolant chacun de ceux-ci à tour
de rôle (toutes les autres substances étant
 6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
1
/
22
100%