Mucoviscidose, CFTR et fonction pancréatique – Cystic fibrosis

177
Métabolismes Hormones Diabètes et Nutrition (XI), n° 4, juillet-août 2007
Nouveaux concepts
Nouveaux concepts
Mucoviscidose, CFTR et fonction pancréatique
Cystic fibrosis, CFTR and pancreatic function
Noélie Davezac*, Aleksander Edelman**
Généralités sur la mucoviscidose
La mucoviscidose est une maladie autosomale récessive qui
affecte approximativement une naissance sur 2 500, majo-
ritairement dans la population caucasienne. Cependant, la
fréquence peut varier sensiblement dans des groupes spéci-
fiques : en France, de 1/2 500 dans le Nord-Ouest à 1/10 000
dans le Sud-Est. Les affections atteignent principalement
les cellules épithéliales de l’appareil respiratoire, du tractus
intestinal, du pancréas, des glandes sudoripares et du tractus
génital. L’affection pulmonaire et l’insuffisance pancréa-
tique sont les manifestations cliniques les plus importantes.
L’atteinte pulmonaire aboutit à des infections pulmonaires
chroniques engendrant des détresses respiratoires qui sont
la cause majeure de la mort des patients atteints d’une
fibrose cystique (CF pour cystic fibrosis) [1]. Environ
90 % des patients souffrent d’une insuffisance du pancréas
exocrine qui entraîne une mauvaise absorption des nutri-
ments et donc une malnutrition (2). Un tiers des patients
ont une fonction hépatique anormale, secondairement à une
infiltration lipidique qui peut évoluer en cirrhose. Plus de
95 % des hommes sont stériles par azoospermie liée à une
absence congénitale des canaux déférents.
Avec l’allongement de la survie des patients, l’intolé-
rance au glucose et le diabète associé à la mucoviscidose
(diabète CF) sont devenus les complications secondaires
les plus fréquentes et pourraient avoir un impact impor-
tant sur la gestion de la maladie.
Le gène CFTR : principalement impliqué dans la
mucoviscidose
Le gène impliqué dans la mucoviscidose a été identifié en
1989 (3). Le gène CFTR (cystic fibrosis transmembrane
conductance regulator) est un gène de 190 kilobases codant
une protéine de 1 480 acides aminés appelée protéine
CFTR (4). CFTR est un canal composé de 12 domaines
transmembranaires impliqué dans le transport d’ions chlo-
rure sur la surface apicale des cellules épithéliales.
On compte à l’heure actuelle plus de 1 400 mutations réper-
toriées (www.genet.sickkids.on.ca/cftr/). Les mutations se
répartissent en cinq classes. Les mutations de classe 1 sont
liées à un défaut de production de la protéine CFTR ; il y
a donc peu ou pas de canaux chlorure fonctionnels. Les
* CNRS FRE 2937, Villejuif ; université Paris-XI, faculté des sciences d’Orsay,
Orsay.
** Inserm U806, Paris.
La mucoviscidose est la maladie autosomale
récessive létale la plus répandue dans la population
caucasienne.
Le gène CFTR (cystic fibrosis transmembrane
conductance regulator) est directement impliqué dans
la pathogenèse de la mucoviscidose et code un canal
transporteur d’ions chlorure.
Plus de 1 500 mutations ont été recensées, et réper-
toriées en cinq classes.
Le canal chlorure CFTR est situé au niveau de la
membrane apicale des cellules épithéliales.
Les principales atteintes de la mucoviscidose
concernent les poumons, le tractus digestif, le foie, le
pancréas et le tractus génital.
Les atteintes pancréatiques concernent directe-
ment le pancréas exocrine, dans lequel CFTR joue le
rôle de fluidifiant du suc pancréatique.
Avec l’allongement de la durée de vie des
patients atteints de mucoviscidose, on observe une
augmentation de la fréquence du diabète associé à
la mucoviscidose (diabète CF), qui présente certaines
caractéristiques.
L’apparition d’un diabète CF est un facteur de
mauvais pronostic pour le développement de la
maladie.
Les pancréatites chroniques ont pour nombre
d’entre elles une origine inconnue (idiopathique).
Au sein de populations atteintes de pancréatites
chroniques idiopathiques, des mutations ont été
observées dans les gènes PRSS1, SPINK1 et CFTR.
▲
▲
▲
▲
▲
▲
▲
▲
▲
▲
points FORTS

178
Métabolismes Hormones Diabètes et Nutrition (XI), n° 4, juillet-août 2007
Nouveaux concepts
Nouveaux concepts
Na+
Na+
K+
HCO3
-
C1-
C1-
CFTR
+
AMPc
Acinus
Sécrétions
acineuses
(riches en protéines)
Suc
pancréatique
(alcalinisation
et dilution)
Canal excréteur
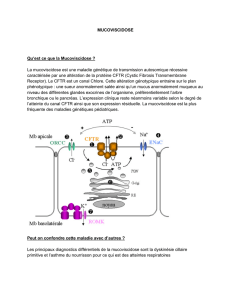
Figure. Modèle proposé pour le rôle de CFTR pendant la sécrétion
pancréatique.
mutations de classe 2 impliquent un défaut de maturation de
CFTR ; les canaux mutés ne peuvent atteindre la membrane
plasmique où leur fonction est requise. Les mutations
de classe 3 touchent la régulation de CFTR, même si la
protéine est capable de s’ancrer dans la membrane. Les
mutations de classe 4 aboutissent à un défaut de conduc-
tance. Enfin, la cinquième classe de mutations engendre
une synthèse réduite de CFTR fonctionnel.
Cette revue sera essentiellement axée sur les atteintes
pancréatiques de la mucoviscidose et l’implication de
CFTR dans les fonctions pancréatiques.
La mucoviscidose :
une atteinte du pancréas exocrine
Quelques données cliniques
L’insuffisance du pancréas exocrine est présente chez
environ 90 % des patients atteints de mucoviscidose. Cette
atteinte semble résulter d’un volume réduit de sécrétions
pancréatiques avec de faibles concentrations en ions bicar-
bonates (HCO3-). Sans une quantité suffisante de fluides et
d’ions HCO3-, les proenzymes digestives sont retenues dans
les canaux pancréatiques et prématurément activées, abou-
tissant à la destruction du tissu et à la fibrose. La mauvaise
absorption des nutriments qui en résulte contribue à ce que
l’énergie nécessaire pour combler les demandes causées
par l’infection bronchique fasse défaut. Ce facteur peut
exacerber l’infection pulmonaire, aboutissant à un cercle
vicieux de malnutrition et d’infection. Les patients présen-
tant une insuffisance pancréatique sont supplémentés en
vitamines liposolubles et en enzymes pancréatiques.
Modèle de fonctionnement de CFTR au sein du
pancréas
Un modèle de fonctionnement de CFTR pendant la sécré-
tion pancréatique a été proposé en 1991 (figure) [5]. Le
suc pancréatique, riche en protéines et sécrété par les
acini, est dilué et alcalinisé lors de son passage au niveau
des canalicules. CFTR intervient au niveau apical des
cellules épithéliales bordant les plus petits canalicules
du pancréas. À cet endroit, CFTR contrôle la sécrétion
d’ions bicarbonates (HCO3-) médiée par l’AMPc (AMP
cyclique) en fonctionnant comme canal anionique (Cl-) et
comme régulateur des échanges HCO3-/ Cl-. La sécrétion
anionique dirige le flux de sodium (Na+) [au travers des
jonctions serrées] et d’eau dans la lumière du canalicule.
Des niveaux plus bas de CFTR aboutissent à une dimi-
nution de la sécrétion de bicarbonates et de fluides. Par
conséquent, un dysfonctionnement de CFTR à ce niveau
peut engendrer la formation d’agrégats protéiques et
prédisposer ainsi le pancréas à des lésions (6).
Le diabète : une complication majeure
de la mucoviscidose
Plusieurs études ont estimé la prévalence du diabète
associé à la mucoviscidose : 5 à 50 % de patients sont
concernés selon les études, auxquels s’adjoignent 15 à

179
Métabolismes Hormones Diabètes et Nutrition (XI), n° 4, juillet-août 2007
Nouveaux concepts
Nouveaux concepts
40 % de patients atteints d’intolérance au glucose (7-
9). Ces fortes différences sont le reflet de la variété des
origines ethniques, des diverses tranches d’âge et de la
diversité des méthodes d’investigation. Le diabète associé
à la mucoviscidose apparaît vers l’âge de 20 ans (9-11),
mais, au-delà de l’insuffisance du pancréas exocrine et de
l’avancée en âge, aucun paramètre clinique ou biologique
n’a pu être identifié comme prédicteur de développement
d’une intolérance au glucose ou de l’apparition d’un
diabète associé à la mucoviscidose.
La mucoviscidose est caractérisée par une mauvaise
absorption/digestion des graisses, un catabolisme
protéique accru et une augmentation des dépenses éner-
gétiques significative. Ces caractéristiques induisent un
traitement particulier, notamment en termes de gestion
de la diététique. Le régime alimentaire reposera sur une
nourriture riche en graisses (35 à 40 % de l’énergie) et en
protéines (15 à 20 % de l’énergie), représentant 120 % des
apports caloriques habituellement recommandés (12).
Caractéristiques cliniques du diabète
associé à la mucoviscidose
Sur le plan clinique, ce diabète présente des similitudes
avec les diabètes de type I et II, mais également de
grandes différences. L’apparition du diabète vers l’âge de
20 ans (diabète de type I avant 20 ans et diabète de type II
bien après 20 ans), un index de masse corporelle bas et
une première phase au cours de laquelle la sécrétion d’in-
suline est faiblement affectée, avec une progression vers
une déficience sévère (nécessitant de multiples injections
d’insuline), sont des particularités du diabète associé à la
mucoviscidose. Comme le diabète de type II, le diabète
CF ne s’associe pas à des stigmates d’auto-immunité
mais, comme lui, il s’accompagne d’un infiltrat amyloïde
(amyline sécrétée en même temps que l’insuline par les
cellules bêta et possédant un effet anti-insuline) et, sur le
plan clinique, d’un risque rare de cétoacidose.
Corrélations entre génotypes et phénotypes ?
Les relations entre génotype et diabète CF sont controver-
sées. Pour certains, le développement d’un diabète chez des
patients atteints de mucoviscidose est corrélé à l’insuffi-
sance pancréatique, elle-même associée à des mutations de
CFTR, principalement la mutation ΔF508 (13-15). Cepen-
dant cette corrélation n’a pas été confirmée par d’autres
(16, 17). De plus, dans la population générale, l’hétéro-
zygotie pour une mutation de CFTR n’est pas un facteur
de risque pour le diabète de type II (18, 19). Finalement,
de rares mutations ont été reliées à un risque augmenté
(N1303K ou W1282X) ou à une absence de risque (A455E)
pour le diabète associé à la mucoviscidose (8, 17).
Influence du diabète sur la progression
de la mucoviscidose
Un certain nombre d’observations suggère que le diabète
CF ne serait pas seulement un marqueur de la sévérité
de la maladie mais aurait intrinsèquement un impact sur
le pronostic de la mucoviscidose. Les patients atteints de
diabète CF ont une survie à l’âge de 30 ans qui est inférieure
à 25 %, alors qu’elle est de 60 % au même âge pour ceux
qui ne développent pas ce diabète (10). Cela converge avec
les résultats provenant de l’American Cystic Fibrosis Foun-
dation Patients Registry, qui annonce une mortalité multi-
pliée par six pour les patients atteints de diabète CF. De plus,
dans un modèle prédictif de survie à 5 ans, le diabète est un
puissant facteur pronostique de décès précoce. Les études
ont montré que le diabète CF est associé à une aggravation
de la fonction pulmonaire et à une altération du statut nutri-
tionnel. La dégradation de la fonction pulmonaire ainsi que
la perte pondérale commencent 2 à 4 ans avant le diagnostic
de diabète CF (20). Le traitement de ce diabète inverse rapi-
dement la perte de poids et la diminution de la fonction
pulmonaire qui lui sont associées (21-23).
Physiopathologie du diabète
associé à la mucoviscidose
Le déficit en insuline est annoncé comme étant la cause
première de diabète CF ; cependant, une résistance à l’insu-
line est également présente chez les patients (1). Le déficit
de sécrétion d’insuline est dû, d’une part, à une réduction
de la masse des cellules β des îlots de Langerhans et à des
anomalies fonctionnelles, d’autre part. Certains travaux ont
démontré une réduction significative du marquage de l’insu-
line à la surface des cellules dans les îlots de patients atteints
de diabète CF comparés à des patients CF non diabétiques
et à des patients contrôles (24, 25). Outre la fibrose, qui est
une caractéristique majeure de la mucoviscidose, il existe
également une infiltration lipidique et un dépôt d’amy-
line dans les îlots, de manière identique à ce que l’on peut
observer dans le diabète de type II. Ces anomalies anatomo-
pathologiques se traduisent par une diminution et un retard
de la sécrétion d’insuline en réponse à des injections intra-
veineuses de glucose ou à son absorption orale (26).
Implication de CFTR
dans les pancréatites chroniques
Quatre-vingts pour cent des pancréatites chroniques sont
causées par l’alcool. La majorité des pancréatites chroni-
ques non alcooliques sont dites “idiopathiques”, c’est-à-
dire d’origine inconnue. Des données récentes suggèrent

180
Métabolismes Hormones Diabètes et Nutrition (XI), n° 4, juillet-août 2007
Nouveaux concepts
Nouveaux concepts
que des altérations génétiques sont des facteurs de risque
avérés pour les pancréatites chroniques. Le risque le plus
élevé est associé à des mutations autosomales dominantes
du gène du trypsinogène cationique (PRSS1). Des muta-
tions ont été identifiées dans le gène de l’inhibiteur de la
trypsine pancréatique (SPINK1) et dans le gène CFTR.
Les mutations du gène CFTR ont été associées à des
pancréatites idiopathiques, mais elles apparaissent égale-
ment comme cofacteurs de l’apparition de pancréatites
chroniques de cause connue (notamment alcooliques).
Une première étude a inclus 134 malades, dont 71 atteints
de pancréatite alcoolique et 60 de pancréatite idiopathique
ou d’une autre origine : 2 hyperparathyroïdies et une
hypertriglycéridémie (27). La fréquence des mutations de
CFTR était de 13,4 %, contre 5,3 % dans la population
témoin. Une autre étude, ne portant que sur 27 patients
atteints de pancréatite chronique idiopathique, a montré
que 37 % d’entre eux possédaient une ou des mutations
de CFTR (28). Ces deux premières études, qui datent de
1998, n’ont recherché respectivement que 22 (27) et 18
(28) mutations du gène CFTR. Depuis, une vingtaine
d’études ont été entreprises et ont objectivé une extrême
variabilité de la fréquence des mutations observées. Les
différences obtenues peuvent s’expliquer par le petit
nombre de patients inclus, l’hétérogénéité des patients
(pancréatites d’origine alcoolique, métabolique, idiopa-
thique ou héréditaire) et enfin par le nombre de mutations
recherchées et la technique utilisée pour les révéler.
Plusieurs études ont entrepris le séquençage complet
de CFTR dans le cadre de travaux sur les pancréatites.
L’étude espagnole a inclus 37 personnes atteintes de
pancréatite alcoolique et 31 atteintes de pancréatite
idiopathique. Les fréquences de mutations retrouvées de
CFTR sont respectivement de 40,5 % (pancréatite alcoo-
lique) et de 38,7 % (pancréatite idiopathique) [29]. Le
travail de Noone et al. a inclus 39 personnes atteintes de
pancréatite idiopathique. Toutes ont eu un screening de
PST1, de PRSS1, de 18 mutations de CFTR, et 20 ont
entièrement été séquencées (16 sujets présentant une
ou des mutations révélées par le screening et 4 présen-
tant apparemment 2 allèles normaux lors du screening).
Vingt-quatre des 39 patients ont des mutations de CFTR.
Les auteurs concluent que le risque de pancréatite est lié
à la présence de 2 mutations de CFTR et à une fonction
réduite de CFTR ailleurs que dans le pancréas. De plus,
la présence de la mutation N34S dans PST1 augmente
le risque de manière indépendante (30). Une équipe
française a recruté 39 patients atteints de pancréatite
chronique idiopathique et a séquencé entièrement le
gène CFTR. Dix-huit allèles anormaux de CFTR ont été
identifiés chez 14 patients. De plus, 20,5 % d’entre eux
(8/39) avaient les mutations les plus communes, ce qui
représente 7,4 fois la fréquence attendue (2,8 %) [31].
En 2005, une étude a montré la présence de mutations ou
de variants de CFTR chez 18 patients sur 40 atteints de
pancréatite chronique idiopathique, confirmant ainsi les
données précédentes (32). Une autre étude axée sur la
recherche de mutations rares de CFTR chez des patients
atteints de pancréatite chronique idiopathique a montré
que le risque de développer cette maladie était forte-
ment lié à l’état hétérozygote du gène CFTR lorsque
l’un des deux allèles est normal. Le risque est multiplié
par quatre dans cette population (33). À l’heure actuelle,
il est donc admis que des mutations de CFTR sont des
facteurs de prédisposition génétique à la survenue d’une
pancréatite (34).
La pancréatite liée aux mutations de CFTR est de patho-
genèse probablement mixte, canalaire et acineuse. L’alté-
ration du canal CFTR est à l’origine d’une déficience de
la sécrétion ductulaire en eau et ions bicarbonates, défi-
cience responsable de précipitations protéiques et d’une
obstruction canalaire (35). Certains travaux ont égale-
ment suggéré une instabilité membranaire dépendante
du pH au niveau des cellules acineuses. La mesure du
potentiel nasal et le test de la sueur s’avèrent normaux
ou peu perturbés chez les patients atteints de pancréatite
et avec une mutation de CFTR, mais sans mucoviscidose
avérée (27, 28, 30).
Conclusion
Le rôle de CFTR dans la fonction pancréatique est direct
au niveau des plus petits canalicules pancréatiques.
CFTR contrôle la sécrétion d’ions HCO3- en fonction-
nant comme canal anionique et comme régulateur des
échanges HCO3-/Cl-. Un dysfonctionnement entraîne
une obstruction des voies exocrines et une destruction
progressive du tissu aboutissant à une fibrose (d’où
le nom de cystic fibrosis employé dans les pays anglo-
américains). Ces observations ont, depuis quelques
années, intéressé la communauté scientifique travaillant
sur les pancréatites idiopathiques. De récents travaux ont
montré une corrélation entre des mutations de CFTR et
l’apparition de pancréatites.
Cependant, les atteintes du pancréas exocrine ne sont
pas les seules pour des patients souffrant de mucovisci-
dose. En effet, avec l’allongement de la durée de vie, un
diabète associé à la mucoviscidose apparaît de plus en
plus fréquemment.
Par conséquent, les relations entre mucoviscidose, CFTR
et fonction pancréatique sont plus souvent étudiées
qu’auparavant et révèlent des corrélations jusqu’alors
ignorées.

181
Métabolismes Hormones Diabètes et Nutrition (XI), n° 4, juillet-août 2007
Nouveaux concepts
Nouveaux concepts
Références bibliographiques
1.
Moran A, Hardin D, Rodman D et al. Diagnosis, screening and management
of cystic fibrosis related diabetes mellitus: a consensus conference report. Dia-
betes Res Clin Pract 1999;45:61-73.
2.
Ratjen F, Doring G. Cystic Fibrosis. Lancet 2003;361:681-9.
3.
Kerem B, Rommens JM, Buchanan JA et al. Identification of the cystic fibro-
sis gene: genetic analysis. Science 1989;245:1073-80.
4.
Girodon-Boulandet E, Costa C. Génétique de la mucoviscidose. Médecine
Thérapeutique Pédiatrie 2005;8:126-34.
5.
Marino CR, Matovcik LM, Gorelick FS et al. Localization of the cystic
fibrosis transmembrane conductance regulator in pancreas. J Clin Invest
1991;88:712-6.
6.
Cohn JA. Reduced CFTR function and the pathobiology of idiopathic pan-
creatitis. J Clin Gastroenterol 2005;39:S70-77.
7.
Moran A. Diagnosis, screening, and management of cystic fibrosis-related
diabetes. Curr Diab Rep 2002;2:111-5.
8.
Mackie AD, Thornton SJ, Edenborough FP. Cystic fibrosis-related diabetes.
Diabetes Med 2003;20:425-36.
9.
Lanng S, Hansen A, Thorsteinsson B et al. Glucose tolerance in patients with
cystic fibrosis: five year prospective study. Br Med J 1995;311:655-9.
10.
Finkelstein SM, Wielinski CL, Elliott GR et al. Diabetes mellitus associated
with cystic fibrosis. J Pediatr 1988;112:373-7.
11.
Riggs AC, Seaquist ER, Moran A. Guidelines for the diagnosis and therapy
of diabetes mellitus in cystic fibrosis. Curr Opin Pulm Med 1999;5:378-82.
12.
Stallings VA. Gender, death and cystic fibrosis: is energy expenditure a
component? J Pediatr 2003;142:4-6.
13.
Solomon MP, Wilson DC, Corey M et al. Glucose intolerance in children
with cystic fibrosis. J Pediatr 2003;142:128-32.
14.
Cucinotta D, De Luca F, Scoglio R et al. Factors affecting diabetes mellitus
onset in cystic fibrosis: evidence from a 10-year follow-up study. Acta Paediatr
1999;88:389-93.
15.
Correlation between genotype and phenotype in patients with cystic
fibrosis. The Cystic Fibrosis Genotype-Phenotype Consortium. N Engl J Med
1993;329:1308-13.
16.
Lanng S, Thorsteinsson B, Erichsen G et al. Glucose tolerance in cystic
fibrosis. Arch Dis Child 1991;66:612-6.
17.
Cotellessa M, Minicucci L, Diana MC et al. Phenotype/genotype correla-
tion and cystic fibrosis related diabetes mellitus (Italian Multicenter Study).
J Pediatr Endocrinol Metab 2000;13:1087-93.
18.
Braun J, Arnemann J, Lohrey M et al. No association between the delta
F508 cystic fibrosis mutation and type 2 diabetes mellitus. Exp Clin Endocrinol
Diabetes 1999;107:568-9.
1. La protéine CFTR est un canal transmembranaire transportant des ions Na+.
2. CFTR est situé au niveau de la membrane apicale des cellules épithéliales.
3. L’apparition d’un diabète associé à la mucoviscidose n’a aucune influence sur le pronostic de la maladie.
4. La mucoviscidose est une maladie génétique autosomale récessive.
Auto-test
Réponses : 1. Faux – 2. Vrai – 3. Faux – 4. Vrai.
19.
Castellani C, Quinzii C, Altieri S et al. A pilot survey of cystic fibrosis clinical
manifestations in CFTR mutation heterozygotes. Genet Test 2001;5:249-54.
20.
Schaedel C, de Monestrol I, Hjelte L et al. Predictors of deterioration of
lung function in cystic fibrosis. Pediatr Pulmonol 2002;33:483-91.
21.
Lanng S, Thorsteinsson B, Nerup J et al. Diabetes mellitus in cystic fibro-
sis: effect of insulin therapy on lung function and infections. Acta Paediatr
1994;83:849-53.
22.
Reisman J, Corey M, Canny G et al. Diabetes mellitus in patients with
cystic fibrosis: effect on survival. Pediatrics 1990;86:374-7.
23.
Rolon MA, Benali K, Munck A et al. Cystic fibrosis-related diabetes melli-
tus: clinical impact of prediabetes and effects of insulin therapy. Acta Paediatr
2001;90:860-7.
24.
Abdul-Karim FW, Dahms BB, Velasco ME et al. Islets of Langerhans in
adolescents and adults with cystic fibrosis. A quantitative study. Arch Pathol
Lab Med 1986;110:602-6.
25.
Robert JJ, Mosnier-Pudar H. Diagnostic et traitement du diabète de
l’adulte atteint de fibrose cystique. Rev Mal Resp 2000;17:798-801.
26.
Costa M, Potvin S, Berthiaume Y et al. Diabetes: a major co-morbidity of
cystic fibrosis. Diabetes Metab 2005;31:221-32.
27.
Sharer N, Schwarz M, Malone G et al. Mutations of the cystic fibrosis gene
in patients with chronic pancreatitis. N Engl J Med 1998;339:645-52.
28.
Cohn JA, Friedman KJ, Noone PG et al. Relation between mutations of the
cystic fibrosis gene and idiopathic pancreatitis. N Engl J Med 1998;339:653-8.
29.
Casals T, Aparisi L, Martinez-Costa C et al. Different CFTR mutatio-
nal spectrum in alcoholic and idiopathic chronic pancreatitis? Pancreas
2004;28:374-9.
30.
Noone PG, Zhou Z, Silverman LM et al. Cystic fibrosis gene mutations and
pancreatitis risk: relation to epithelial ion transport and trypsin inhibitor gene
mutations. Gastroenterology 2001;121:1310-9.
31.
Audrezet MP, Chen JM, Le Marechal C et al. Determination of the relative
contribution of three genes - the cystic fibrosis transmembrane conductance
regulator gene, the cationic trypsinogen gene, and the pancreatic secretory
trypsin inhibitor gene - to the etiology of idiopathic chronic pancreatitis. Eur J
Hum Genet 2002;10:100-6.
32.
Bishop MD, Freedman SD, Zielenski J et al. The cystic fibrosis transmem-
brane conductance regulator gene and ion channel function in patients with
idiopathic pancreatitis. Hum Genet 2005;118:372-81.
33.
Cohn JA, Neoptolemos JP, Feng J et al. Increased risk of idiopathic chronic
pancreatitis in cystic fibrosis carriers. Hum Mutat 2005;26:303-7.
34.
Maire F. Quand demander un bilan génétique devant une pancréatite aiguë
ou une pancréatite chronique ? Gastroenterol Clin Biol 2005;29:715-23.
35.
Choi JY, Muallem D, Kiselyov K et al. Aberrant CFTR-dependent HCO3-
transport in mutations associated with cystic fibrosis. Nature 2001;410:94-7.
1
/
5
100%