Syndrome de Marfan - Swiss Medical Forum

ARTICLE DE REVUE
Syndrome de Marfan
Christine H. Attenhofer Josta, Marianne Rohrbachb, Gabor Matyasc, Florian Schoenhoffd, Matthias Baumgartnerb,
Angela Oxeniuse, Thierry Carreld, Michael Hueblerf, Kan Ming, Gabriella De Pasqualeh, Klara Landaui,
Oliver Kretschmare, Dragan Odavick, Paul Vogta, Francesco Faletral, Matthias Greutmannm
a HerzGefässZentrum Zürich, Klinik Im Park, Zürich; b Stoffwechselabteilung, Forschungszentrum für das Kind, Universitäts-Kinderspital Zürich;
c Zentrum für Kardiovaskuläre Genetik und Gendiagnostik, Schlieren-Zürich; d Herz- und Gefässchirurgie, Inselspital Bern;
e Kardiologie, Universitäts-Kinderspital Zürich, f Klinik für Herz- und Gefässchirurgie, UniversitätsSpital Zürich; g Swiss Scoliosis, Klinik Im Park, Zürich;
h Kardiologie, Stadtspital Triemli, Zürich; i Augenklinik, UniversitätsSpital Zürich; k Klinik für Herzchirurgie, Stadtspital Triemli, Zürich;
l Cardiocentro Ticino, Lugano; m Abteilung Erwachsene mit Kongenitalen Vitien, Klinik für Kardiologie, UniversitätsSpital Zürich
Introduction

ARTICLE DE REVUE
Forum Médical Suisse
Nosologie actuelle
Dilatation aortique et dissection aortique
Tableau 1:
Sans anamnèse familiale positive pour le syndrome de Marfan:
Avec anamnèse familiale positive pour le syndrome de Marfan:
Pas de syndrome de Marfan:

ARTICLE DE REVUE
Score systémique
(striae dis-
tensae)
Tableau 2:
Atteinte systémiquePoints
(striae)
Figure 1:
Figure 2:
Segment
supérieur
Segment
inférieur

ARTICLE DE REVUE
Anamnèse familiale et analyse génétique
Figure 3:
A:
B:
Main normale Syndrome de MarfanSigne du pouceSigne du poignet
AB
Figure 4:

ARTICLE DE REVUE
Echocardiographie, IRM ou tomodensito-
métrie: quel examen à quel moment?
American College of
Cardiology / American Heart Association / American As-
sociation for Thoracic Surgery
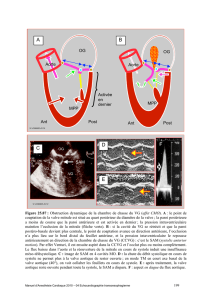
Figure 5:
 6
7
8
9
10
11
12
6
7
8
9
10
11
12
1
/
12
100%